


The next generation of functional materials will need to include aniostropic (directionally dependent) crystals. But how has nature been the source of inspiration for these?
-
Mimicking biological materials can help design the next generation of functional materials
-
Biopolymers are prime candidates for producing aniostropic crystals

The best designs are in nature
Biomimetics is literally the mimicking of nature; taking advantage of the millions of years of evolution which has produced fabulous and functional natural materials using minimal resources and effort and replicating those processes in the laboratory.
In many cases, the materials we use in computing, catalysis, structural engineering and vehicle design can all be improved by synthesizing them in a way which follows the principles and practices seen in nature.
Often stunningly beautiful, but always with a function to perform, living organisms excel in controlling crystal growth, producing the sorts of shapes which can be hard to coax out of crystals growing in a beaker. But there are some very good reasons for us to try ...
A challenge
A challenge faced by scientists is to produce materials for device applications which satisfy the continual drive for higher performance, in much faster time scales.
An example of how this inexorable drive has progressed over the last 40 years is to consider how integrated circuit technology has developed. In 1969 the guidance computer that helped Apollo 11 land on the moon had a 1 MHz clock speed, 4K RAM and weighed 32 kg. Compare this to the iPhone 3GS, which has an 833 MHz clock speed, 16 Gb RAM, weighs only 135 g and fits easily into a pocket. This and other astounding developments have mainly been possible due to improvements in the way scientists command matter.
The materials which most excite research activity have been chosen as they exhibit functionality, either for use as simple conductors1 or for more complex emergent behaviour as UV lasing,2 photoluminescence3,4 or even in a biomedical role as drug delivery facilitators and targeted medical devices.5
Thinking ahead, it is likely that in order to incorporate functional materials directly into the next generation of even smaller and faster electronic devices, anisotropic forms of these materials, eg nanowires, nanotapes, or nanotubes will be required.6
In terms of shape, anisotropic crystals have one or more of the axes significantly different in length than the others. In practice this means that the crystals will be either flat and plate-like, or long and thin like wires.
When using anisotropic materials in devices, the long and wire-like shape is preferred. This is because the key feature of this particular shape of nanomaterial is the confinement of properties such as electron conduction in all dimensions bar one. The in-built inhibition of electron conduction along the short axes provides an important control as adding features such as switches and insulating barriers can be extremely difficult at the nanoscale and will invariably affect its performance.
An excellent review of the fabrication of nanowires7 describes in great detail many of the materials which have been synthesized and which have found application.
The general challenge of synthesizing anisotropic functional materials can be further broken down into key problem areas:
- the complex materials synthesized must be at the nanoscale
- they must be single-phase
- they must be able to be synthesized in large amounts.
As if this wasn't enough, there are the additional requirements through social mores, legislation and economics that the syntheses must ideally be:
- green
- cheap
These demanding criteria effectively rule out the majority of synthetic protocols. Solid-state 'heat and beat' routes produce multi-micron or even centimetre-sized crystals, and the fact that the more complex the material, the greater the possibility for side-reactions forming unwanted associate phases. There are techniques which can produce phase pure materials at the nanoscale, but at the expense of a tiny amount of product for a great deal of effort. Synthetic routes using templates to form nano-sized reaction volumes, such as micelles and vesicles satisfy most of the criteria. However, many surfactants and complexing agents are by-products of the petrochemical industry, so environmental concerns rule them out as being ideal for the drive towards green syntheses.
Nature shows the way

All is not lost however. For the last 500 million years, all of the criteria above have been satisfied successfully and without the hand of man interfering.
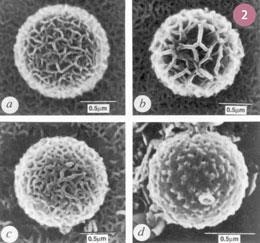
Consider the humble diatom. Diatoms are unicellular algae which possess a bewildering array of complex and often strikingly beautiful skeletons, with structural detail at the nanoscale (fig 1). To synthesize their delicate microscopic bodies, diatoms use proteins and polysaccharides to direct the assembly of silica to follow the shape of the underlying organic phase. The replication is highly species-specific. Driven by the underlying genetic direction, any given species can produce structures on the nano- and micro-scale with perfect reproducibility in extremely large amounts. In some cases, the 'blooms' of billions upon billions of diatoms can be seen from space.
If some in society are worried about the rise of nanobots and the coming of the 'grey goo', they may like to reflect on the fact that these self-replicating, self-sustaining nano-organisms have been quietly existing in the biosphere for the last 160 million years or so.
Biomimetics is a relatively recent area of investigation, yet one which has already had far-reaching implications for the way in which crystal growth control is effected in inorganic systems.
Initial experiments in biomimetics8,9 produced complex and often life-like inorganic materials, by following closely the mineralisation protocols used in nature (fig 2).
The fact that biomimetics works so well is largely due to the replication of the complimentary interaction between the oppositely charged biopolymer and inorganic phase.
Formation of the smallest possible crystal 'nucleus' is a critical determining factor in the growth of an inorganic material, a process which is invariably slow and will lead to 'classical' crystal morphologies (cubes, rhombs, prisms, etc.). Organic matter however, serves as a fully flexible, charge complimentary substrate for the formation of nuclei. This means that crystallization will occur wherever the charge-matching is most favourable.
Given that these sites of nucleation are determined by the overall structure of the organic phase, the crystal shapes which can be generated are virtually limitless. With a greater number of nucleation events occurring on the organic substrate, crystallite size is kept naturally very small, thereby leading to a more precise replication of the organic matter by the inorganic material.
Biomimetic synthesis
The first biomimetic syntheses were concerned with producing complex structures using the same inorganic phases encountered in nature, such as calcium carbonate, silica and hydroxyapatite. These are all exceedingly useful materials, largely though in a structural role, providing skeletal support for micro- and macro-organisms. The chemistry of these materials is very well known and relatively simple. It is difficult to arrive at anything other than calcium carbonate when the reagents are those used in equation 1.
Na2CO3 + CaCl2 → 2NaCl + CaCO3 (1)
Many examples of biomimetic syntheses of these 'simpler' materials exist and the reader is directed to a very good review.10
Pertinent to the quest for anisotropy in more complex functional matter, such as semi-conductors, catalysts or superconductors, is the fact that useful anisotrophic shapes of some simple materials have been achieved through biomimetics, using biopolymers such as starch, chitosan from crab shells and dextran from bacteria.
Starch is a ubiquitous glucose polymer and what makes it particularly important for this work is that it is inherently anisotropic at the molecular scale, being comprised of long polysaccharide chains. This anisotropy can be transferred easily to materials introduced into starch solutions. Nanowires of polypyrrole are one such example.11
Polypyrrole is a simple polymer constructed from pyrrole heterocycles, which was found to have exceptionally high electrical conductivity. Polypyrrole nanowires are produced by adding pyrrole to a solution of starch. This results in adsorption of the pyrrole onto the starch via hydrogen bonding. The adsorbed pyrrole monomers can then be polymerised in situ, the polymerisation following the chains of starch molecules to form nanowires. Electron microscope images of the material reveal a uniform wire-like nanostructure with average diameter of approximately 100 nm and lengths varying from hundreds of nanometres to several microns.
A tough test
When one tries to apply biomimetics to the synthesis of more chemically complex materials however, things aren't so straightforward. The problems of limiting unwanted side-reactions and controlling crystal growth can be illustrated in the synthesis of a superconductor YBa2Cu2O7-δ(Y123) which is superconducting at the relatively high (for a superconductor) temperature of around 90 K.
To achieve the correct crystal phase of Y123, the synthesis demands a temperature of 900°C which is well outside the realm at which proteins and polysaccharides are viable as templates to directly control mineralisation. In addition, the potential for side-reactions in this system is huge. Heating the correct stoichiometric amounts of yttrium, barium and copper with a simple polysaccharide such as starch can lead to the formation of a product which could contain BaCO3, BaO, Y2 BaCuO5, CuCO3, CuO, Y2 O3, Cu2 Y2 O5 and many other stable crystal phases, none of which is Y123.
What is required therefore, is a biotemplate which can effectively sequester all the precursor cations, keep the system homogeneous and prevent unwanted side reactions from happening until the only favourable reaction is the one which gives the desired product. Such a seemingly impossible set of criteria was recently found to be satisfied by what is at first glance a very simple biomolecule, alginate.
Alginate is a polysaccharide found in brown seaweeds such as the kelp Laminaria. Chemically, the biopolymer is an unbranched, anionic polysaccharide, consisting of blocks of polyguluronate (-G-)n, polymannuronate (-M-)n and alternating (-G-M-)n residues, the proportions of which depend on the species of seaweed from which the alginate is extracted. This simple di-block structure gives rise to a complexity which the seaweed uses to great effect.
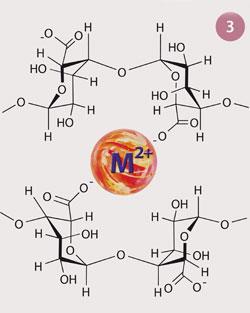
The ability of poly-G blocks to bind cations strongly is an emergent feature of the molecular shape of these blocks, which is particularly pronounced when multiple alginate molecules are in close proximity.
When two poly-G regions on separate alginate molecules align, a pocket is created in which mechanically strengthening cationic cross-linking can occur readily (fig 3).
Within a particular species of seaweed, the amount and position of the poly-G and poly-M blocks vary, depending on the structural properties that the alginate must give to the plant. As it is the poly-G blocks which strongly sequester cations such as Na+ from the seawater, the regions of the seaweed which need to be the most resistant to tidal forces (eg the holdfast and stalk), will be rich in poly-G residues, whereas the fronds which require flexibility to billow in the tide, will be richer in poly-M residues. As the poly-G regions are regularly spaced in any given alginate, this leads to a regular array of cross-linking junction zones, the so-called 'egg-box' model of ion binding. It is in these regions that scientists are able to swap Na+ for virtually any other cationic species.
The introduction of metal cations to a solution of alginate will result in preferential uptake of the metallic species into the poly-G regions. As the uptake ability of these regions is so strong, there is virtually no distinction between cations of different elements. This ensures that there is essentially atomic homogeneity. The potential for side-reactions with regions of one cationic species concentrated together has immediately been substantially reduced.
Taking again the superconductor Y123, once the Y3+, Ba2+ and Cu2+ are introduced to a solution of alginate and heating begins, the first crystal phase to form is barium carbonate. With the barium perfectly sequestered within the egg-boxes, these crystals remain nano-sized.
Once past 300°C, the alginate starts to carbonise, forming barium carbonate nanoparticles and preventing them from growing any larger. This leaves arrays of well dispersed nanoparticles throughout the amorphous carbon/yttrium/copper matrix.
As 811°C is reached, the barium carbonate nanoparticles begin to melt and can then act as catalytic sites for the uptake of copper and yttrium ions from the amorphous matrix. Nanowires of the desired superconducting phase then grow radially from the barium carbonate nanoparticles.12
Scanning electron microscopy (SEM) reveals that the superconducting material is comprised of fibrous aggregates, up to several microns thick (fig 4). Using a transmission electron microscope (TEM), it was found that the fibres themselves are comprised of bundles of straight-sided nanowires, typically 10 nm in diameter.

Confirmation of the superconducting properties of these materials was by way of superconducting quantum interference device (SQUID) magnetometry, which showed that these nanowires had a critical temperature of 77 K, which is only slightly lower than the 85 K-89 K typical for Y123.
Now that this remarkable biopolymer has been demonstrated to successfully yield nanowires of a complex oxide superconductor, scientists are finding that it works for other functional materials too.
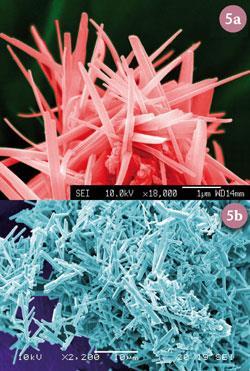
Using similar synthetic techniques, alginate has been used to produce nanowires of LSMO (La0.67Sr0.33MnO3) a complex manganite used in magnetic data storage applications (fig 5a) and langasite (La3 Ga5 SiO14), a lead-free piezoelectric material (fig 5b). In the case of the latter, until alginate was used, all attempts at controlling crystal growth in this material had failed, owing to the extremely high favourability of competing side-reactions. The success of these syntheses rely on the complex emergent behaviour of alginate in the adoption of egg-box zones of high metal-cation chelating ability.
It is only a matter of time before a similar, or even more complex behaviour in another biopolymer is identified and utilised.
Conclusion
In order to fulfill the challenges outlined at the beginning of this article, biopolymers are clearly prime candidates for a number of reasons.
Cocooning metal salts in biopolymers ensures that the inorganic phase will remain nano-sized. If it is required, the biopolymer can provide a ready source of carbon during synthesis. This could be used either to form metal carbonate or carbide phases or to ensure that the reaction environment is a reducing one.
Most biopolymers, particularly polysaccharides, are composed of long molecular chains, which means that anisotropy is 'hard-wired' into any organic/inorganic cooperative crystal growth process.
Finally, perhaps the most important attributes that a biopolymer brings are that they are on the whole extremely cheap and environmentally benign. The two most abundant biopolymers, cellulose and chitosan are produced in gigaton amounts worldwide either naturally by plants and animals, or as by-products of the food and construction industries.
It may be another decade or two before anisotropic nanomaterials find their way into the latest generation of devices, but the ever-evolving role of biomimetics in the synthesis of these materials will ensure that when the device is ready to be built, a suitable single crystal nanowire of any desired functional material will be to hand.
Related Links
International weekly journal of science
Initial experiments in biomimetics
Initial experiments in biomimetics
References
- D A Wharam et al, J. Phys. C: Solid State Phys., 1988, 21, L209 (DOI: 10.1088/0022-3719/21/8/002)
- M H Huang et al, Science, 2001, 292, 1897 (DOI: 10.1126/science.1060367)
- M S Gudiksen et al, J. Phys. Chem. B, 2002, 106, 4036 (DOI: 10.1021/jp014392n)
- H-Y Chen et al, Opt. Express, 2008, 16, 13465 (DOI: 10.1364/OE.16.013465)
- L A Bauer et al, J. Mater. Chem., 2004, 14, 517 (DOI: 10.1039/B312655B)
- Z L Wang, Adv. Mater., 2003, 15, 432 (DOI: 10.1002/adma.200390100)
- K S Shankar and A K Raychaudhuri, Mater. Sci. Eng., C, 2005, 25, 738 (DOI: 10.1016/j.msec.2005.06.054)
- D Walsh and S Mann, Nature (London, U. K.), 1995, 377, 320 (DOI: 10.1038/377320a0)
- S Mann, Angew. Chem., Int. Ed., 2000,39, 3392
- S Mann and G A Ozin, Nature (London, U.K.), 1996, 382, 313 (DOI: 10.1038/382313a0)
- W Shi et al, Chem. Commun., 2007, 2414 (DOi: 10.1039/B701592E)
- Z A C Schnepp et al, Adv. Mater., 2008, 20, 1782 (DOI: 10.1002/adma.200702679)


No comments yet