


When nuclear magnetic resonance (NMR) was described more than half a century ago it appeared to be a curiosity of the quantum world. Since then NMR spectroscopy has become an essential tool not only for chemists, but also for biochemists, molecular biologists and even clinicians. In the first of three articles to highlight the power of this technique, the virtues of one dimensional NMR are explored.
-
Observed by physicists in the 1930s, the nuclear magnetic resonance effect has been exploited by chemists in structural analyses for decades
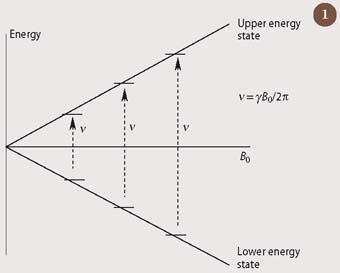
Nuclear magnetic resonance (NMR) spectroscopy involves the excitation of nuclei by electromagnetic radiation in the radio-frequency range of the electromagnetic spectrum. For a nucleus to absorb energy from radiowaves in this way, it must have the quantum mechanical property of spin. A spinning nucleus, such as that of the hydrogen atom, will adopt one of only two possible states when placed in a magnetic field. (In NMR, the hydrogen nucleus is often referred to as a proton, and is given the abbreviation 1H.) As the strength of the magnetic field is increased, there is a proportional increase in the energy 'gap' between these two states, as is shown in Fig 1. We can predict the resonant frequency at which any spinning nucleus will absorb energy from radio-frequency radiation (ν), as it jumps from the lower energy state to the upper state, by using the fundamental equation:
ν = γB0/2π (i)
where B0 is the magnetic field which the nucleus experiences, and ν is a proportionality constant, which is specific to that nucleus.
Physicists explore nmr
The first scientists to investigate nuclear magnetic resonance were physicists whose primary concern was to measure fundamental physical constants. One of the first magnetic resonance experiments was done in 1938 by the Noble prize-winning physicist, Isidor Rabi, while studying molecular beams to measure nuclear magnetic moments (the magnetic moment is related to the constant γ in equation (i)). Soon after this, the American physicist, Felix Bloch succeeded in measuring the magnetic moment of the neutron, again using a beam of particles (this time, polarised neutrons, subatomic particles with the property of spin). Fortunately, these early researchers also investigated less exotic forms of matter. Indeed Bloch and fellow American physicist, Edward Mills Purcell, won the Nobel prize for physics in 1952 for demonstrating that substances such as water and paraffin could also be used to observe nuclear magnetic resonance.

The equation ν = γB0/2π was easily solved for these liquid samples - water (H2O), for example, contains only one kind of hydrogen atom and therefore produces a single resonant frequency. However, it soon became apparent that this simple equation did not fully describe the 1H-nmr phenomenon for most molecules that were typically studied in solution. Both chemical structure and sample preparation subtly impinge on the absorption process shown in Fig 1. The power of one dimensional NMR derives from our understanding of the origins of these minute effects and from our ability to relate these small changes, when observed, to the underlying chemistry of the sample. We will explore three of the most useful factors for chemists - chemical shift, coupling and the nuclear Overhauser effect.
The chemical shift
Nuclear magnetic resonance first came to the attention of chemists following the discovery that protons in different chemical environments resonate at very slightly different frequencies. Thus, a molecule which contains hydrogen in different chemical environments will present not just one radio-frequency absorption, as is the case for water, but rather a spectrum of 1H resonances. For example, two clearly distinct peaks appear in the 1H-nmr spectrum of lactic acid, which is shown in Fig 2. Each peak corresponds to a different chemical environment for hydrogen in this molecule.
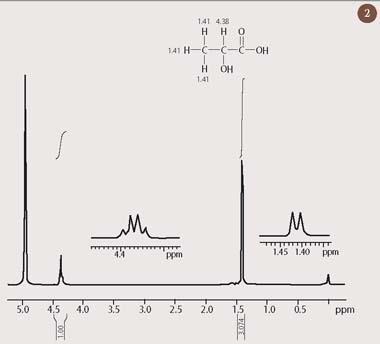
This tiny displacement in the resonant frequency of a nucleus - which has come to be referred to as its 'chemical shift' - turns out to be a direct probe of local chemical structure. The chemical shift results from minute differences in the external magnetic field which is experienced by each nucleus. These local differences are, in turn, a consequence of small additional magnetic fields which are produced by the electrons in the vicinity of the nucleus. The 1H chemical shift therefore reflects the local electronic environment for a hydrogen nucleus within a molecule. It is possible to associate a characteristic proton chemical shift to a particular functional group with a fairly high degree of certainty. (Fig 3 shows approximate ranges for some common proton environments.) One of the great strengths of NMR spectroscopy is that the presence of more than one functional group results in an 'additive' - and therefore predictable - change to the chemical shift.
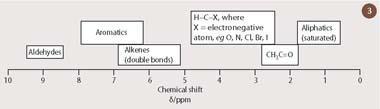
We can now confidently assign the peak at 1.41 ppm in Fig 2 to the hydrogen atoms in the methyl group of lactic acid, because saturated aliphatic protons, such as these, normally resonate within a narrow range of chemical shifts, between 0.5-1.5 ppm (Fig 3). This is a consequence of the relatively high density of electrons at the hydrogen atom of such an unpolarised C-H bond. By contrast, a proton which is attached to a carbon atom that also bears an electronegative element will routinely appear in a distinct, but equally well-defined region of the 1H-nmr spectrum, between 3-5 ppm (see Fig 3). Thus, the methine C-H bond in lactic acid, for which the carbon atom is substituted by an electronegative oxygen atom, is strongly polarised towards the oxygen substituent. Consequently, there is a reduced electron density at this hydrogen atom, resulting in its appearance at 4.38 ppm in the 1H-nmr spectrum of lactic acid (Fig 2).
Chemists were quick to realise that analysis of the chemical shifts in a 1H-nmr spectrum could be used to list the different chemical environments in a molecule, in much the same way that infrared (IR) spectroscopy had been used to identify functional groups. One of the many advantages of 1H-NMR spectroscopy, as compared to IR, is that it provides an accurate indication of the relative number of protons in each chemical environment. This is achieved by integrating the area under each peak in the spectrum, an operation which is easily done by the same computer that controls the nmr spectrometer. The resulting 'integral' is displayed as a line above the peak, rising by an amount which is proportional to the area of that peak. Thus, inspection of the heights of the two integrals in the 1H-nmr spectrum of lactic acid (Fig 2) reveals that there are approximately three protons resonating at 1.41 ppm for every one proton at 4.38 ppm (the precise relative values of these integrals, 3.074:1.000, are also displayed underneath the chemical shift scale). This confirms the assignments of these peaks to the CH3 and CH groups, respectively. Such quantitative information could not have been obtained easily from an infrared spectrum.
Coupling
As well as feeling the effects of nearby electrons, nuclei can also interact with other spinning nuclei, through the chemical bonds which connect them. This weak interaction is referred to as 'coupling' and it leads to the phenomenon of 'peak splitting' which is observed in many 1H-nmr spectra. For example, inspection of the two expansions in the spectrum of lactic acid (Fig 2) reveals that the three-proton group at δH 1.41 is split into two peaks, while the single-proton resonance at δH 4.38 is split into four. There are various ways of explaining such peak splitting in terms of the coupling interaction between neighbouring nuclei. For example, the 'n + 1' rule states that whenever a proton is coupled to n equivalent 1H nuclei, it will be split into an n + 1 'multiplet'.
Coupling is most commonly observed between hydrogen nuclei that are connected to one another by three chemical bonds, as is the case for the protons in the methyl and methine groups of lactic acid. Thus, the single proton at 4.38 ppm is coupled to three protons in the adjacent methyl group, and is therefore expected to be split into four peaks by application of the n + 1 rule. The methyl group at 1.41 ppm experiences the same coupling interaction with its single-proton neighbour, and this causes the methyl group to be split into two (1+1). These splitting patterns for lactic acid are referred to as a 'quartet' and a 'doublet' respectively, and this pattern of splitting is characteristic for any system comprising a group of three chemically equivalent protons which is coupled to a single-proton neighbour.
The n + 1 rule permits splitting patterns to be predicted for many different arrangements of coupled hydrogen nuclei within a molecule. Effectively, therefore, the splitting patterns observed in a 1H-nmr spectrum can be used to make 'connections' between sets of nuclei, through the intervening chemical bonds. This understanding of splittings has turned out to be one of the most powerful features of NMR spectroscopy, since it is possible to use the information to 'piece together' the structure of an unknown compound in a way which would be impossible to achieve by any other spectroscopic technique.
Pulsed-FT NMR spectroscopy
The first 'continuous wave' (CW) nmr spectrometers, which became commercially available in the 1960s, worked in a similar way to the infrared spectrometers of the time. The frequency of the electromagnetic radiation used in excitation (radiowaves) could be varied while maintaining a constant magnetic field (or vice versa) to bring each proton-containing chemical moiety sequentially into resonance. In this way, CW-nmr spectrometers provided an unrivalled view of the location of hydrogen atoms in a molecule. However, the usefulness of NMR was to take a quantum leap in the early 1970s as a result of two technological innovations.
The first of these was the capability for providing simultaneous excitation of the whole spectrum of hydrogen frequencies which are present in a molecule, by using a short pulse of radio-frequency energy. The second was the application of high-speed computers to disentangle all the resulting superimposed signals by means of a mathematical operation, a Fourier transform (FT), which then produced a 'normal-looking' nmr spectrum.2 As a result of the vastly more efficient use of time inherent in the new pulsed-Fourier transform technique, it became possible to record nmr spectra from nuclei which had previously been impossible in practice, either because of their low natural abundance or because of their low resonant frequency (ie low γ in Fig 1). (1H was the first nucleus to have been observed spectroscopically because it is present at almost 100 per cent natural abundance and because it has the highest resonant frequency of any stable isotope in the Periodic Table.)
The new pulsed-FT technique could be used to study the isotope carbon-13. Unfortunately, carbon-12, which is the more abundant isotope of carbon, does not have a nuclear spin and is therefore 'invisible' in nmr. Since 13C represents only ca 1 per cent of the total carbon in most samples, the improved efficiency of the pulsed-FT technique was essential for its detection in any realistic sample. Pulsed-FT 13C-NMR spectroscopy is now second only to 1H-NMR spectroscopy in its importance for chemical research.
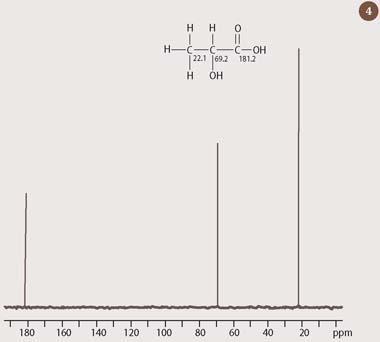
A 13C-nmr spectrum provides the organic chemist with a direct means of counting the number of carbons in a molecule. More significantly, however, the 13C chemical shift responds to variations in the local chemical environment in much the same way as 1H does, albeit over a wider range. The 13C chemical shift scale runs from 0 to 200 ppm, and it is therefore possible to estimate the chemical shift for most carbon atoms, provided that the shift of the corresponding proton is known, simply by multiplying by 20 the appropriate ranges for proton (between 1-10 ppm in Fig 3). Thus, in the 13C-nmr spectrum of lactic acid (Fig 4), the CH carbon resonates at 69 ppm - a little less than 20 times the value of the corresponding methine proton. Similarly, the CH3 carbon, at 22 ppm, falls within the range 10-30 ppm, which would be predicted from multiplying the corresponding aliphatic (saturated) hydrogen values in Fig 3 by 20. (The third peak at 180 ppm comes from the carbon of the carboxylic acid group.)
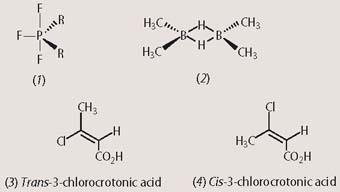
Using the new pulsed-FT technique, chemists could record an nmr signal from almost every element in the Periodic Table, provided that an isotope with the appropriate nuclear spin was available. For example, the trigonal bipyramidal compound (1) presents two 19F resonances in 19F-NMR spectroscopy. These occur with relative intensities in a ratio of 2:1, corresponding to the two fluorine atoms in an axial environment (vertical bonds) and the single fluorine atom in an equatorial environment (horizontal bond), respectively. (In the actual 19F-nmr spectrum, the axial fluorine peaks are split into a doublet of doublets by coupling both to the equatorial 19F and to 31P; while the equatorial fluorine is split into a doublet of triplets by coupling to phosphorus and to both of the axial fluorine atoms.) By contrast, 11B-NMR spectroscopy of the diborane complex (2) detects only a single resonance because the two boron atoms are in identical chemical environments (again, in the actual 11B-nmr spectrum, coupling between 11B and the two bridging hydrogens leads to the boron peak being split - this time into a triplet).
Studies of the isotopes of nitrogen and phosphorus with the appropriate nuclear spin (15N and 31P), have allowed important new applications of NMR spectroscopy to biology and medicine. These include investigations of the solution structure of proteins using 15N-labelled samples; and the diagnosis of clinical disorders, such as tumours and congenital metabolic abnormalities, by in vivo 31P-NMR spectroscopy.
In addition, pulsed-FT NMR permitted the investigation of another 'complication' to the simple model of nmr shown in Fig 1: the nuclear Overhauser effect (nOe).2,3 The nOe is a 'relaxation' phenomenon, associated with the way in which a nucleus releases the energy that it has absorbed during excitation. It turns out that any spinning nuclei which is close in space to another can promote the relaxation of its partner. To perform the nuclear Overhauser experiment, the nucleus of interest is first irradiated at its own specific frequency before applying the radio-frequency pulse in the pulsed-FT experiment. This pulse then excites all the other nuclei simultaneously, and the nOe effect is subsequently observed in the nmr spectrum as an increase in the intensity of all the peaks associated with nuclei which are close in space to the spin that was irradiated at the start of the experiment.
The size of the observed nOe gives us information about the distance between these nuclei, which is independent of the interactions occurring through chemical bonds (as such, it needs to be carefully distinguished from coupling). For example, irradiation of the alkene proton (δH 6.1 ppm) in trans -3-chlorocrotonic acid (3) results in a nuclear Overhauser enhancement of around 10 per cent at the methyl protons (δH 2.2 ppm), which reside on the same face of the double bond. However, no such increase in peak intensity is observed when the same experiment is repeated for the cis isomer of 3-chlorocrotonic acid (4), in which these two groups are located on opposite sides of the double bond and are therefore further apart. (The 1H-nmr spectra for both isomers of 3-chlorocrotonic acid are almost indistinguishable, with the alkene proton and the methyl group substituted at the double bond appearing at nearly identical positions within their expected chemical shift ranges (δH 5-7 and δH 2-3, respectively, see Fig 3.) Also, there is no peak splitting information in the 1H-nmr spectrum to assist in assigning either spectrum to its appropriate isomer. Under these circumstances, the nuclear Overhauser effect is the only tool capable of resolving the issue of stereochemistry around this particular double bond.
One dimensional NMR - the future

To this day, the main drawback to high-resolution NMR spectroscopy is its poor sensitivity when compared with other techniques of analytical chemistry, such as UV-VIS spectroscopy and mass spectrometry, both of which require very much less sample. On the plus side, however, NMR spectroscopy is a non-destructive technique so the sample can be recovered at the end of the experiment.
Over the past 50 years, the manufacturers of nmr spectrometers have sought to address this problem through technological advances. As a result, the size of the available magnetic field has increased by more than 15 times: from the first permanent magnets, with a 1H resonance frequency of 60 MHz, to a modern-day 950 MHz super-conducting magnet, operating at a temperature close to absolute zero.
The improvements in magnet technology have been a measure of progress, but there have also been parallel, though less visible, improvements in the hardware used for radio-frequency excitation, which have resulted in equally dramatic improvements in the sensitivity of an nmr instrument. The most recent such innovation has been the introduction of cryogenically-cooled probes.4 In these systems, the electronics responsible for receiving the radiofrequency nmr signal are cooled to a temperature close to that of liquid helium, resulting in a up to four-fold increase in the sensitivity of the nmr experiment. This sizeable leap in sensitivity is revolutionising many areas of high-resolution NMR spectroscopy, not least of which is the emerging science of 'metabonomics'.
An nmr-based metabonomics study sets out to measure the concentrations of many metabolites from a biofluid which has been isolated from the human body, such as blood or urine. The 1H-nmr spectra that are recorded in a typical study contain thousands of peaks, representing hundreds of compounds. Many such spectra must be obtained to provide an accurate picture of the biological variation across the population under investigation. Because of the size and complexity of metabonomic studies it is impossible to identify every single resonance from every spectrum. The most favoured approach to extracting meaningful information from the hundreds or even thousands of spectra employed in a metabonomics study is currently to perform a multivariate statistical analysis, using a computer (Fig 5).
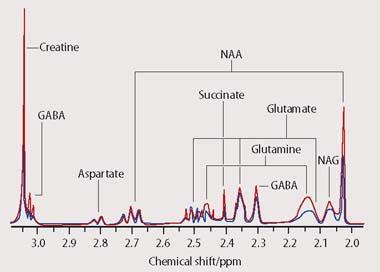
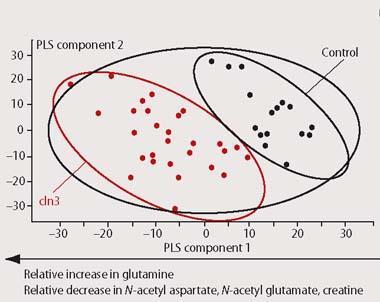
In effect, rather than determining the structure of every component of the biofluid, we produce a 'metabolic profile' of the whole. By visualising the overall 'pattern of metabolism' in this way we can obtain an indirect picture of the various metabolite fluxes occurring in a living organism. Any change in the appearance of the 1H-nmr spectrum from a biofluid will therefore reflect one of many different kinds of possible changes to metabolism within the body. These can include the presence of diseases, which often produce a characteristic fingerprint of biochemical perturbations. As a result both of the increasingly high sensitivity and high sample throughput available from modern nmr spectrometers, metabonomics is now coming to be an increasingly valuable tool for diagnosis in clinical medicine.5
Dr Geoff Brown is lecturer in NMR spectroscopy in the department of chemistry at the University of Reading, PO Box 224, Whiteknights, Reading RG6 6AD.
References
1. A. Abragam, The principles of nuclear magnetism. London: OUP, London. 1961.
2. R. R. Ernst, G. Bodenhausen and A. Wokaun, Principles of nuclear magnetic resonance in one and two dimensions. Oxford: Clarendon, 1987.
3. T. D. W. Claridge, High-resolution NMR techniques in organic chemistry. Tetrahedron Organic Chemistry Series, vol 19. Amsterdam: Elsevier, 2004.
4. H. Kovacs, D. Moskau and M. Spraul, Prog. Nuc. Mag. Res. Spect., 2005, 46, 131.
5. J. C. Lindon, E. Holmes and J. K. Nicholson, Prog. Nuc. Mag. Res. Spect., 2004, 45, 109.



No comments yet