


How do we detect cancer markers in the blood, cocaine in a mixture where it has been deliberately masked, and pollutants in drinking water? Electronic biosensors based on DNA hold the answer.
-
Chemists exploit the ability of DNA to recognise complementary DNA sequences in the development of medical and environmental sensors

There are many types of sensors but all rely on a molecular recognition event, such as antibody-antigen binding, which can be coupled to a read-out device, a number on an LCD display for example. Ideally, a sensor should be highly specific and as portable and easy to use as a home pregnancy kit - simply dip the sensor into the liquid and read the result. However, few sensors achieve this level of perfection. A promising new approach is to use DNA sequences which have been selected to bind certain targets (DNA aptamers) and link them to an electronic read-out device via an electrochemical circuit.
Early gene sensors
The idea of DNA-based sensors was pioneered in the early 1990s by Kevin Plaxco at the University of California at Santa Barbara, and Ciara O'Sullivan at the Universitat Rovira i Virgili i Tarragona, Spain. There is a precedent in Nature, as protein scientists had come to realise, that many proteins remain unfolded in a cell until they encounter a target molecule at which point they grab the target by folding around it, resulting in a highly specific fit.
Plaxco started by investigating DNA because nucleic acids are easier to synthesise and handle in the laboratory than proteins. He developed a gene sensor- E-DNA -which relied on the ability of DNA to switch between alternative conformations. The immobilised sensor DNA folds up on itself in a hairpin conformation. However, if it encounters the target gene, which offers a larger number of stable base-pair interactions, the DNA strand stretches out and binds the target instead.
In such a device (see Fig 1) the sensor DNA is bound to a gold surface at one end, while the other end carries a ferrocene label, which serves as an electrochemical signal. The DNA includes a sequence complementary to the target DNA sequence, and a stretch of self-complementary sequence, forcing it to fold up in a hairpin structure unless the target sequence binds to the strand. While the DNA is buckled up in the hairpin conformation, the ferrocene stays so close to the gold surface that electron transfer is possible from the ferrocene to the surface. When the sensor DNA finds and binds its target sequence it stretches out, moving the ferrocene label away from the surface and suppressing the signal.1
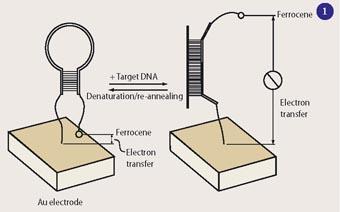
Earlier DNA sensors based on optical or fluorescence detection required a spectrometer to read the results. The E-DNA is smaller and could be developed into a portable electronic device. E-DNA is highly specific and recognises differences in a single base. The initial experiments were done with pure solutions of DNA targets in buffer. Recently, however, Plaxco's group demon-strated that E-DNA also works well with complex mixtures such as blood, food, and soil samples.2
DNA aptamers
The ability of DNA to recognise complementary sequences is just one of the many things that this molecule can do. With the toolkit of modern molecular biology, producing large amounts of a given piece of DNA has become straightforward. Since the 1990s, researchers have used these tools to create large libraries of random sequences and then identify and mass-produce specific sequences with desirable binding properties. Such DNA binders, known as aptamers, serve as unchangeable molecular recognition elements, like a piece of a jigsaw that will fit only with the specific neighbouring pieces.
Plaxco and his coworkers set out to fit aptamers into their sensors. They reasoned that by replacing the gene probe with a known DNA aptamer, they should be able to create an electronic sensor for the aptamer's target, or indeed for any target against which a DNA aptamer could be found. Their first aptamer sensor was based on a known aptamer specific to the blood-clotting enzyme thrombin. The sensor could detect this protein directly in blood serum (ie blood minus the red and white blood cells) down to a limit of around 20 nanomolar.3
While this sensor successfully demonstrated the feasibility of these new sensors, it had a minor blemish. Like the first version of the E-DNA sensor, it was constructed in such a way that the appearance of the target molecule suppresses an existing electrochemical signal (seeFig 2 (a)). Thus, it gives a 'negative' signal. In this sensor, a single DNA strand is attached to a gold electrode and carries both the aptamer sequence and the methylene blue (MB) label. The strand on its own is flexible, so that MB will be near the surface at least some of the time and engage in electron transfer. Binding of the target makes the DNA strand fold up in a rigid conformation which moves the MB away from the surface and suppresses the signal. For a sensor to have maximum sensitivity and specificity, the molecular recognition event should trigger a 'positive' signal.
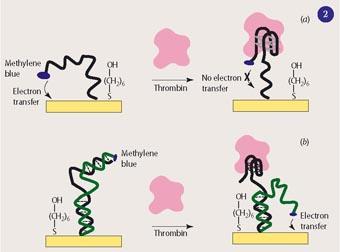
Ciara O'Sullivan and her coworkers were the first to build an electronic sensor, with DNA aptamers, with a positive signal. This was also for detecting thrombin, but owing to a subtle difference in the construction of the DNA probe, it achieved the more desirable positive mode straightaway.4
By contrast, Plaxco's group had to tweak the DNA constructs to turn their sensors into positive versions of both the thrombin5 and the E-DNA6 sensors (see Fig 2 (b)). To this end they introduced a second DNA strand, complementary to the one bound to the gold surface, which ensures that the label stays away from the surface. Binding of the target displaces this DNA and allows the label to approach the surface. The extra effort paid off - this thrombin sensor turned out to be 10-fold more sensitive in the positive mode. Technically, this was because the new sensor exhibited higher gain (ie a larger change in current at a given target concentration).
Into the real world
In a first step towards real applications, Plaxco's group developed a DNA aptamer-based sensor for detecting cocaine in blood serum. The sensor could be used even in the presence of masking substances commonly used in drug trafficking,7 and turned out to be several orders of magnitude more sensitive than the Scott test. (A colorimetric-based test, routinely used at border controls, which is easily obscured by mixing cocaine with dark substances.)
Remarkably, two pupils from the local high school in Santa Barbara, who joined Plaxco's group for a work experience placement took part in developing the cocaine sensor. They built their own specimen, contributed measurements used in the paper, and were rewarded by appearing as co-authors.
In another project, Plaxco's group developed an aptamers-based sensor for PDGF (platelet-derived growth factor), a potentially important indicator for cancer. The device can pick up the clinically relevant PDGF concentrations in the picomolar range directly from the serum.8 Direct comparison of these aptamer-based electronic sensors with optical or fluorescence-based detectors showed that they performed equally well as the others in sensitivity, but outperformed them in target specificity when analysing blood samples.
Plaxco's group has also used a DNAzyme, ie a piece of DNA with catalytic activity, instead of an aptamer. They developed a sensor for lead based on a known DNAzyme which depends on lead ions for its activity. Like aptamers, DNAzymes can be identified by screening large numbers of random sequences, and if a requirement for a metal cofactor is desired, one would include that metal in the screening.
The architecture of this sensor is similar to the aptamer sensors: one end of the DNA is immobilised onto a gold surface, the other carries an electrochemical label. The metal-free DNAzyme forms a stable double helix with the substrate DNA, thus the label doesn't get near the surface. Only when the DNAzyme binds a lead ion will it cut and release the substrate DNA, so that it is left behind as a flexible single strand and the label moves nearer the surface and an electrochemical signal appears.8
The detection limit of this sensor is 62 ppb (parts per billion), which is well below tolerable levels of lead in environmental and in clinical analysis. The US Food and Drug Administration (FDA), for example, has set the limit for lead in products used by children to 500 ppb, while a recent EU directive limits the lead content in electronic components to 1000 ppb. The researchers tested a range of other bivalent ions, including Mn, Ni, Zn, Co, Cu, and Hg and found no cross-reaction. Since the ionic radius of Pb2+ (1.20Å) is larger than that of common metal ions, any metal ion that might cross-react would be rarer than lead and probably just as undesirable.
Outlook
First tests with electronic biosensors based on DNA aptamers have shown that they come close to being the ideal sensor. For some applications, however, it may become necessary to introduce additional checks and balances to exclude other factors influencing the rearrangement of the DNA. O'Sullivan, for example, admits that her thrombin sensor is as sensitive to changes in potassium ion concentration as to thrombin concentration. Nevertheless, it appears that electronic aptamer-based sensors offer significant advantages over other types of biosensors and that they will establish themselves in a range of applications.
References
- C. Fan et al, Proc. Natl. Acad. Sci. U.S.A., 2003, 100, 9134.
- A. A. Lubin et al, Anal. Chem., 2006, 78, 5671.
- Y. Xiao et al, Angew. Chem. Int. Edn, 2005, 44, 5456.
- A.-E. Radi et al, J. Am. Chem. Soc., 2006, 128, 117.
- Y. Xiao et al, J. Am. Chem. Soc., 2005, 127, 17990.
- Y. Xiao et al, Proc. Natl. Acad. Sci. U.SA., 2006, 103, 16677.
- B. R. Baker et al, J. Am. Chem. Soc., 2006, 128, 3138.
- R. Y. Lai et al, Anal. Chem., 2007, 79, 229.
- Y. Xiao et al, J. Am. Chem. Soc., 2007, 129, 262.



No comments yet