


The history of pharmaceuticals is enriched by accounts of drugs developed for one therapeutic purpose that found application in another. This is true for chlorpromazine, a treatment for severe mental illness
The history of pharmaceuticals is enriched by accounts of drugs developed for one therapeutic purpose that found application in another.1 This is true for chlorpromazine, a drug that led to a revolution in the treatment of severe mental illness, particularly schizophrenia.

Schizophrenia is a debilitating illness that usually becomes evident during adolescence or early adult life and will affect some 7-8 individuals per 1000 during their lifetime. Most people think of this illness as causing hallucinations and delusions (false beliefs that others do not share). While true, many of the other difficulties faced by people with this condition are a result of altered patterns of thinking that make many everyday tasks very difficult, particularly those involving planning and problem-solving, and a related loss of motivation and drive.
Modern medication is quite effective at controlling the hallucinations and delusions, but much less effective at helping with the other symptoms. Until the early 1950s, treatment was limited and patients would spend years, sometimes even the rest of their lives, in hospital. The literature from around 1950 focused on the place of electroconvulsive treatment, insulin coma treatment and psychoanalytical interpretations, with passing reference to trials of cortisone, deoxycortisone and ascorbic acid. None was particularly effective and 'treatment' consisted more of custodial management than therapy. The advent of chlorpromazine changed the whole environment in the psychiatric hospitals - almost overnight, control of symptoms and recovery were realistic goals.
Antihistamine start
Chlorpromazine started life as an antihistamine, one of a group of drugs used for treating allergic reactions. The first recorded fatal allergic reaction caused the death of King Menses of Egypt who, sometime between 3640 and 3300 BC, died from a wasp sting, having been sensitised by an earlier sting. Over five millennia later, in 1902, Paul Portier and Parisian physiologist Charles Richet coined the term 'anaphylactic shock' to describe this type of reaction. They were trying to protect dogs against the poisonous tentacles of sea anemones, and injected the animals with an extract prepared from the tentacles. A fortnight later the dogs were re-injected, in anticipation that some form of immunity would have been induced. The animals died, suffering from the classic symptom of 'shock' - a sudden and dramatic fall in blood pressure. Rather than being protected from the poison, they had been sensitised to it. A little later medics John Auer and Paul Lewis, at the Rockefeller Institute for Medical Research, New York noticed 'hypersensitivity' in guinea pigs when they were given a second injection of protein. Typically, there would be a sudden constriction of the bronchioles, leading to death.
(Sir) Henry Dale, working in the Wellcome Research Institute on the chemicals occurring in the toxic fungus ergot and those that arose in the course of its putrefaction, noticed a similarity between these reported symptoms and those of histamine poisoning, a substance he had isolated from ergot. In 1910 he drew attention to 'a possible significance that the immediate symptoms with which an animal responds to a normally inert injection of protein to which it has been sensitised are to a large extent those of poisoning by β-iminazolylethylamine (histamine)'.
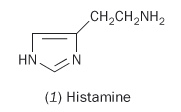
And indeed, this compound, histamine (1) even in minute amounts, has profound physiological effects: a one in 100,000 solution applied to slightly abraded skin produces a fluid-filled wheal (ridge on the skin) as the capillaries leak plasma. Soon histamine was implicated in a range of debilitating diseases, including asthma and many allergies. Clearly development of a drug that could counteract its effects - an antihistamine - would be of great benefit.
Dale's work in 1914 gave the first indication that such a drug might exist. When he injected one of his ergot extracts into a cat, he found the classic symptoms of shock were caused by acetylcholine in the extract rather than histamine. Excitingly, the shock induced by acetylcholine could be blocked by an injection of atropine. This suggested that release of histamine might also be suppressed by an appropriate antagonist. Three tests were developed so that a number of chemicals could be screened for antihistamine activity:
- protection against an otherwise lethal injection of histamine;
- protection against asthma-like symptoms induced by inhaling a histamine aerosol;
- reducing the spasms induced in isolated guinea-pig intestines on treatment with histamine.
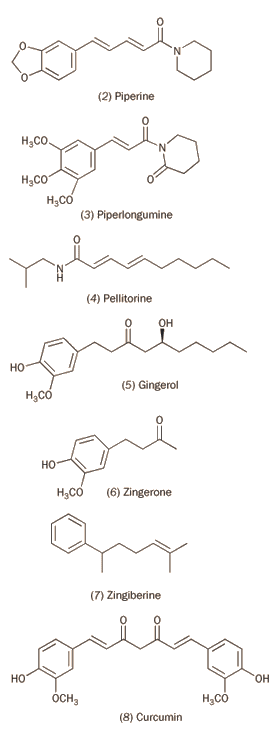

Rhône-Poulenc researcher Daniel Bovet used these screening tests to discover that certain aromatic ethers containing a tertiary amine group had promise (see, for example, compounds 2 and 3) but the first trial compounds proved too toxic for human use. Further development led, in 1947, to Benadryl (4), also known as Acrivastine, which is still in use today to relieve the symptoms of hay fever and nettle rash.2
Useful though these early antihistamines were, at therapeutic doses they induced such marked drowsiness that they interfered with people's everyday living. This provided the incentive for chemists to investigate other classes of drugs for antihistamine activity.
One of the most common agricultural insecticides before World War II was lead arsenate, but it was toxic to humans as well as insects. As early as 1934 the US Department of Agriculture was screening a raft of organic molecules to find one that would kill pests without poisoning people. More or less immediately they found that thiodiphenylamine (phenothiazine, 5) was pleasingly toxic against the test insects, mosquito larvae. This suggested that phenothiazine derivatives might be valuable in the fight against malaria.
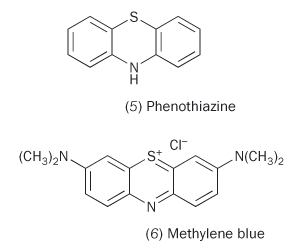
Paul Ehrlich had already demonstrated that the dyestuff methylene blue (6) (an oxidised phenothiazine) could kill malaria plasmodia without harming the human host. However, experiments with 'un-oxidised' phenothiazines led to the conclusion in the 1944 Journal of the American Chemical Society that these drugs were ineffective against the plasmodia. Pierre Viaud, director of pharmaceutical research at Rhône-Poulenc takes up the story:
but unacquainted as we were with (this) publication, we continued the research programme and naturally obtained the same results as this author: the products had no appreciable anti-parasitic action. However, the systematic study of their pharmalogical properties soon revealed that we had an interesting lead ... The antihistaminic activity of some of these products attracted our attention and straightway they were more fully investigated.3
Parkinson's potential
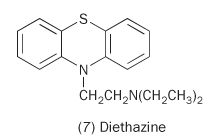
Like Benadryl and its relatives, these phenothiazines made the patients drowsy. The Rhône-Poulenc researchers recognised that this drowsiness meant that the drug was crossing the blood-brain barrier and wondered if they could exploit this. At the time they were seeking agents to help sufferers from Parkinson's disease and one particular phenothiazine derivative (diethazine, 7) showed promise.
Diethazine was initially tried out on 13 patients, 11 of which showed 'good results':
however one must not think that all the symptoms disappeared completely and the change was total; nevertheless, the modification is usually spectacular, and the patients find that the result is superior to that obtained from other treatments, even when well-managed.
As well as the anti-Parkinsonian effect, diethazine and its relatives had an effect on patients' mood. Depression, often associated with the illness, was replaced with feelings of euphoria. So far, so good. But was it possible to proceed further and develop other reagents that would have even more beneficial effects on the central nervous system?
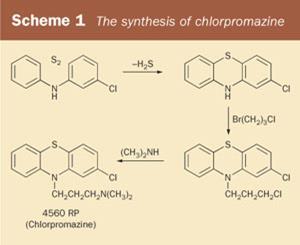
Two screening tests were developed. In the Macht test, rats were trained to climb a rope to feed from a platform at the top. Drugs affecting the CNS would alter the average time the rats took to get to their feeding platform. Both Benadryl and diethazine caused dose-related confusion in the rats, which then affected the time it took to get to their food. But for this story, the second screening test is more relevant. This was to see if the drugs could prolong hexobarbital-induced sleep in mice. Essentially, it tested if the barbiturate hexobarbital was being 'potentiated' by the test drug. They found that one of the phenothiazine derivatives showed exceptional promise. This was compound 4560RP, the 'RP' indicating its connection to Rhône-Poulenc. This phenothiazine derivative was synthesised (see Scheme 1) by chemist Paul Charpentier who later christened it chlorpromazine. The drug didn't help patients suffering from Parkinson's disease - rather, it could exacerbate their symptoms. And this is where a new chapter in this story begins.
Anaesthetic 'potentiator'
Until the mid-1950s one of the safest anaesthetics for major surgery was diethyl ether. However, ether anaesthesia was a significant stress on the body and was considered to contribute to 'surgical shock'. When anaesthetic burden was added to other demands, particularly severe blood loss from war injuries, this could be fatal. In addition, administered by mask, it produced a sense of suffocation that compounded patients' fears. From about 1880, chemists and physicians had searched for other drugs to use during or before anaesthesia to reduce both anxiety in patients and the amount of diethyl ether required for surgery. These 'potentiators' of anaesthetics included morphine, tribromoethanol and scopolamine. None was totally satisfactory and the search for better agents continued until well after World War II.
In 1951 Henri Laborit, a 37-year-old surgeon working in the Val-de-Grâce military hospital in Paris reasoned that 'surgical shock' might be lessened if patients' metabolic rates were reduced by external cooling by using ice packs and administering a mixture of various 'potientiating' drugs. Laborit decided to use antihistamines as part of this cocktail, not for their benefits on allergies such as hay fever, but to exploit their main adverse side effect - a drowsiness, sometimes allied to feelings of drunkenness. He approached the pharmaceutical firm of Rhône-Poulenc for recently developed antihistamines with which to experiment. They gave him a sample of the experimental drug 4560RP, chlorpromazine.
Laborit found that chlorpromazine not only potentiated anaesthesia, but also had another remarkable effect. Instead of patients anxiously awaiting the mask, he noted they showed a certain 'uninterest' in their surroundings and predicaments, which he termed 'euphoric quietude'. He perceptively recognised the potential for this effect in other areas of medicine, noting in his report: 'These findings allow one to anticipate certain indications for the use of this compound in psychiatry, possibly in connection with barbiturates, in a deep sleep cure'.4 ('Deep Sleep Therapy' involved putting patients into a coma lasting between one and eight weeks. Complications of such prolonged unconsciousness could cause death, but it was considered at the time to offer some hope for patients with severe illnesses for whom there was little other treatment.)
By the time his paper was published, three of his colleagues, psychiatrists Joseph Haman, Jean Paraire and Jean Velluz, were trying out 4560RP on their patients. They described the effects on a patient suffering from mania:
The patient is calm after injection (and) sleeps. He wakes up manic again. Over the next three weeks he receives chlorpromazine in combination with analgesic, barbiturate and electroconvulsive therapy. (Then) he was calm enough that he could play bridge and lead a normal life, though his symptoms remained somewhat hypomanic.
This report had three consequences. First, it undermined previous concerns about the safety of the drug arising from the experiences of a single subject, since the drug clearly had not harmed the patient; secondly it stimulated research to ascertain which aspects of the therapeutic regime were responsible for its dramatic effect; and thirdly it provoked, at least among Parisian psychiatrists, realisation that chemical treatment for otherwise intractable mental conditions might be within reach. Trials with chlorpromazine alone were first started by Jean Sigwald and Daniel Bouttier at the Hospital Paul Brousse. Unfortunately they delayed publication until 1953 so that they could report on a larger group of patients. This allowed their rivals Jean Delay and Paul Deniker at the Ste-Anne Mental Hospital to publish first and thus gain fame for being the 'first' to demonstrate the benefits of chlorpromazine in psychiatry. Medical historian Edward Shorter summarises the position:
In May 1952 at a meeting of the Medical Psychiatric Society, they [Delay and Deniker] described their work on chlorpromazine omitting any reference to Laborit. Then at the Society's June meeting they delivered a fuller report on 8 patients. Whilst it is true that they were the first to treat a series of patients exclusively with chlorpromazine it is not true that they discovered the drug's psychiatric applications. And history has wrongly exalted Delay and Deniker in connection with opening this epochal new chapter in the annals of psychiatry, the advent of pharmapsychotherapy, whilst sliding in silence over that of Laborit.5
The early 1950s saw many reports of the effectiveness of the drug, typically:
It calmed restless, excited or overactive patients without sedating them to the level where they could not function. Patients lost their agitation but not their consciousness. They could talk about themselves and eat and sleep without difficulty (the) excitement was no longer life-threatening.
Heinz Lehmann, at the Verdun Protestant Hospital in Montreal reported on its effects on (initially) 71 schizophrenic patients:
Two or three of the acute schizophrenics became symptom-free. Now I had never seen that before. I thought it was a fluke - something that would never happen, but anyway, there they were. At the end of 4 or 5 weeks there were a lot of symptom-free patients. By this I mean that a lot of hallucinations, delusions and disorders had disappeared. In 1953 there wasn't anything that ever produced something like this - a remission of schizophrenia in a matter of weeks.6
Interestingly, there were to be further uses for chlorpromazine in other areas of medicine later - including relieving a wide range of drug- (especially chemotherapy) or disease-induced vomiting, as part of the treatment of tetanus and, perhaps most curiously, in relieving intractable hiccups. While these other uses were 'happened upon', advances in pharmacology today mean that drug actions are increasingly predictable from the pattern of their interaction with the body's receptors, allowing an earlier awareness of additional uses for new drugs.
Alan Dronsfield is professor of the history of science in the school of education, health and sciences at the University of Derby, Kedleston Road, Derby DE22 1GB. Peter Ellis is professor of psychological medicine at the Wellington School of Medicine and Health Sciences, University of Otago, PO Box 7343, Wellington South, New Zealand.
Acknowledgements
Judith Swazey provides an excellent and comprehensive account of the discovery of chlorpromazine and (up to ca 1970) its use in therapy.3 Most of the unattributed quotations in this article come from this source. Edward Shorter supplies a more condensed version of the historical events and includes details of pre-chlorpromazine treatments for schizophrenia and mental illnesses.6
Future treatments for schizophrenia?
Discovery of the effectiveness of chlorpromazine in treating schizophrenia was serendipitous. Subsequent studies suggested it worked by blocking dopamine receptors, particularly the D2 dopamine receptor. While this may be a critical action of most antipsychotic medications, it is not necessarily the only mechanism. Indeed, clozapine, often effective when all previous treatments have failed, binds only weakly to the D2 receptor. (Clozapine can damage the bone marrow, so is used as a last resort.) The dopaminergic and serotonergic systems are closely interrelated, with serotonin suppressing dopaminergic activity. Most of the newer anti-psychotics have some action on serotonergic receptors, and this may account for their side effect profile. Indeed, blocking certain serotonergic receptors may account for the superior effectiveness of clozapine.
Phencyclidine (PCP) is a drug of abuse that can produce a psychosis very similar to schizophrenia by blocking another brain receptor, the NMDA (N-methyl-d-aspartate) receptor. This is present through much of the brain and is crucial to controlling its early development and plasticity. In addition, one of the genes thought to be involved in vulnerability to schizophrenia produces a protein that may regulate the interaction of the NMDA receptor with its ligand, glutamate.7,8
There is no evidence of alteration of the NMDA receptor in schizophrenia. Indeed, a change in such a central part of brain signalling would have profound effects on the individual from before birth.7 However, downstream effects of NMDA receptor activation may account for some symptoms of schizophrenia, particularly the inability to consider, reflect and plan how to solve a problem.9 The amino acids glycine and d-serine can modulate the action of the NMDA receptor. These both produced significant benefit in double-blind, placebo-controlled clinical studies when added to conventional antipsychotics but can be difficult to take. Instead, new drugs might alter mechanisms regulating brain levels of glycine, such as the glycine and small amino acid transporter mechanisms. Clozapine may act, in part, through these mechanisms. Other targets for future treatment may include the recently described brain transporters for d-serine and a group of receptors influencing binding of glutamate to the NMDA receptor.7 Intriguingly, even steroids, one of the drug treatments mentioned in the survey of treatments from 1950, is being re-examined.10 It is now apparent that the brain produces steroids that have direct effects on receptor function, including the NMDA receptor. This offers another avenue to explore for more effective and less burdensome treatments for this challenging disorder.
References
- A. T. Dronsfield and P. M. Ellis, Educ. Chem., 2001, 38 (6), 123.
- G. M. Dyson and P. May, May's chemistry of synthetic drugs, 5th edn. London: Longmans, 1959.
- Quoted in J. P. Swazey, Chlorpromazine in psychiatry. Cambridge (Mass): MIT, 1974.
- H. Laborit, P. Huguenard and R. Alluaume, Presse Med., 1952, 60, 206.
- E. Shorter, A history of psychiatry. New York: John Wiley & Sons, 1997.
- H. Lehmann: part of an interview by Dr David Healy, and reproduced in Ref 5.
- B. Moghaddam, Neuron, 2003, 40, 881. 8. M. J. Owen, N. M. Williams and M. C. O'Donovan, J. Clin. Invest., 2004, 113, 1255.
- D. M. Barch, Psychopharmacology, 2004, 174, 126. 10. W. T. Trost and C. E. Marx, Int. Drug Ther. Newsl., 2005, 40, 1.


1 Reader's comment