


About 10 per cent of men and women over 65, and nearly half of those over 80, have Alzheimer's disease. With the number of people in the world aged over 65 expected to increase by an extra billion by 2050, effective medicines must be developed for this debilitating illness.
- Efficacy achievable with current drugs on market for AD patients is at best modest
- Cholinergic receptor molecules are proving to be better targets for drug development in the treatment of AD
- Drugs to slow or stop AD are now a realistic prospect

In 1906 at a psychiatric clinic in Munich, Alois Alzheimer, a 42-year old German doctor, looked down his microscope at brain tissue from a 51-year old woman (Frau Auguste D) who had died in a local mental asylum following a number of years of progressive dementia. He observed structures previously described in the brain of elderly people - neuritic or senile plaques - alongside structures contained within brain cells that had not been previously observed - neurofibrillary tangles. Both of these microscopic changes are used to characterise the form of dementia that is now known as Alzheimer's disease (AD).
A positive diagnosis
For most of the 20th century, a positive diagnosis of Alzheimer's disease was restricted to a person suffering with dementia who was less than 65 years old. Dementia in older (senile) individuals was regarded as a consequence of vascular disease associated with old age. However, the meticulous studies of two British researchers, John Corsellis, working at Runwell Hospital in Essex in 1962, and Bernard Tomlinson at Newcastle University in 1970, independently demonstrated the presence of plaques and tangles in older individuals in the absence of such degeneration. Thus, although structural changes are more severe and less variable in the presenile form of AD, there was no reason to maintain a distinction on the basis of age. The term 'Alzheimer's disease' now describes dementia of the Alzheimer type irrespective of age of onset. A definitive diagnosis does, however, require demonstrating the presence of plaques and tangles, which can only be done by microscopic examination of brain tissue post-mortem. Before death, the best a psychiatrist can say is that a person probably has AD if they have a history of progressive dementia.
Just over 100 years on since Alzheimer made his discovery, what advances have been made in understanding this 'peculiar disease of the cerebral cortex',1 as he called it, and what are the prospects for new medicines for treating this debilitating illness, which leaves sufferers with severe disorientation and memory loss?
A basis for drug therapy
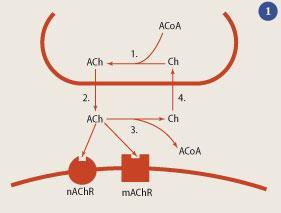
Attempts to treat AD were in the dark ages for more than half of the 20th century. Hydergine, a mixture of egolanine mesylates derived from a fungus that grows on rye, was used to treat dementia despite the fact that its efficacy was uncertain and its mechanism of action unknown. A breakthrough came with the discovery, in the mid-1970s, by David Bowen in London, which was rapidly corroborated by Peter Davies in Edinburgh and Elaine Perry in Newcastle, that the activity of the enzyme responsible for the synthesis of the neurotransmitter acetylcholine (ACh), choline acetyltransferase, was reduced in the cerebral cortex of subjects with AD (see Box).2 This finding rapidly led to the hypothesis that Alzheimer's occurs as a consequence of dysfunction of cholinergic nerve cells (neurons), which established the conceptual framework for the emergence of therapies to enhance cholinergic function.
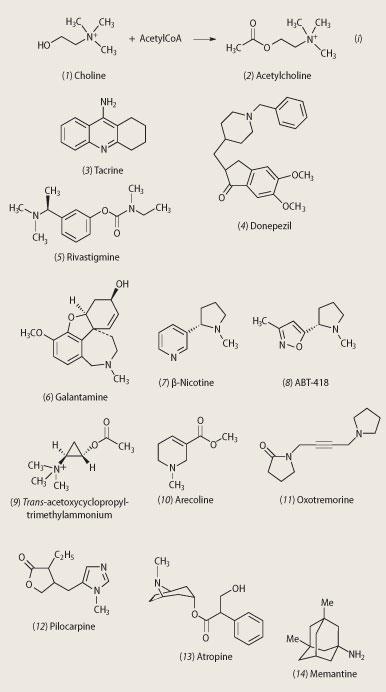
Dietary choline and phosphatidylcholine serve as the sources of free choline (1) for acetylcholine (2) synthesis (see equation (i)). However, attempts to increase cholinergic function by administering metabolic precursors for acetylcholine failed to show efficacy. Such an approach also gave some rather unpleasant side effects. Choline breaks down in the stomach to trimethylamine, the compound responsible for the stench of rotting fish. Another, less malodorous, approach was needed.
Upon release, acetylcholine is metabolised into choline and acetate by the enzyme acetylcholinesterase (AChE), and other non-specific esterases. Inhibition of these enzymes thus provides a potential way of increasing the amount of ACh in the synapse, helping to restore cholinergic function in Alzheimer patients (see Fig 1). The first AChE inhibitor to show clinical activity was tacrine (3) (Cognex, Warner Lambert). In the early 1990s, its assessment constituted the first large-scale multi-centre trial of a drug to treat dementia but the emergence of side effects, specifically liver damage, led to the end of the trial, and an end to tacrine.
However, this work stimulated a search for second generation AChE inhibitors with fewer side effects and longer half-lives. (The half-life of a drug is the time taken for its concentration in the blood to reduce by a half.) The first of these was donepezil (4) (Aricept, Pfizer/Eisai), in 1997, closely followed by rivastigmine (5) (Exelon, Novartis). In 2000 galantamine (6) (Reminyl, Jannsen-Cilag/Shire) was approved for the treatment of mild to moderate AD. However, the efficacy achieved with all of these AChE inhibitors is modest and is not evident in many patients. More specific approaches to cholinergic therapy, ie targeting cholinergic receptors, was necessary.3,4
Nicotinic and muscarinic receptors
Neurotransmitters interact with specific receptor molecules (proteins). In the case of acetylcholine, these are nicotinic and muscarinic receptors - so called because they were originally distinguished on the basis of their selectivity to nicotine and muscarine, respectively. Activation of these receptors by released ACh continues the process of signal transmission.
Most peripheral acetylcholine receptors (AChR ) are nicotinic, such as those on the heart or at the neuromuscular junction, which is why nicotinic antagonists (a drug that blocks nicotinic receptors, such as d-tubocurarine, from the arrow poison, curare) are used as muscle relaxants. Nicotinic agonists (nicotine mimicking drugs), including nicotine (7), have been found to improve attention of Alzheimer patients. Another nicotinic agonist, ABT-418 (8), displays cognition-enhancing properties.
Scientists are currently investigating novel nicotinic agonists which are selective for particular nicotinic receptor subtypes. A number of such compounds are in clinical development, many of which are less toxic than nicotine.
Muscarinic receptors are stimulated by muscarine, and blocked by atropine, which is the poison (also known as deadly nightshade) found in the belladonna plant. Muscarinic receptors have been divided into five receptor subtypes. Several synthetic muscarinic agonists have been made, based on the structure of acetylcholine. Research points to the three-membered ring of acetoxycyclopropyl-trimethylammonium iodide having the highest intrinsic activity; the trans -isomer (9) having much higher activity than the cis-isomer. Such information will be important in the development of new drugs. To date, however, potent muscarinic agonists - eg arecoline (10), oxotremorine (11) and pilocarpine (12) - have been found to be of limited therapeutic value in AD because of the wide range of cholinergic responses that they elicit. Oxotremorine, for example, evokes tremors and thus provided an early model for Parkinson's disease.
Muscarinic antagonists such as scopolamine (also known as hyoscine) and atropine (13) are among the oldest known molecules. Both induce memory loss in humans, which support a role for cholinergic neurotransmission in memory.
A new approach
The possible use of antagonists to muscarinic receptors located on cholinergic nerve terminals to increase the concentration of acetylcholine in the synapse has become a major focus recently. Muscarinic autoreceptors (ie muscarinic receptors that modulate the release of ACh) are similar to a muscarinic receptor found in the heart. Studies have shown that blocking such receptors increases the extracellular concentrations of ACh in the cortex. The muscarinic antagonist AF-DX-116, for example, has been shown to improve cognitive performance in experimental animals. A number of compounds from Schering-Plough, including SCH-217443, have shown efficacy in increasing ACh release and improving cognitive function in rats. Alvameline, a combined muscarinic agonist and antagonist, was investigated as a potential treatment for AD, but development was discontinued following the completion of phase II/III trials because of failure to demonstrate therapeutic efficacy.
As well as the presence of plaques and tangles, AD is characterised by a progressive loss of pyramidal cells. These neurons use excitatory amino acids (mainly aspartate and glutamate) as the neurotransmitter and play a vital role in the mediation of excitatory synaptic transmission. Receptors for these amino acids, for example the N -methyl-d-aspartate (NMDA) receptor, are also thought to play a key role in both cognitive function and neurodegeneration. Memantine (14) is an example of an NMDA receptor antagonist which was launched onto the market in Europe in 2002 by Merz (as Axura) and Lundbeck (as Ebixa), and in the US in 2004 by Forrest.5 The drug represents the world's first and only medication for the treatment of moderate to severe AD (the AChE inhibitors on the market are licensed for mild to moderate AD only).
Although the cause of the characteristic, selective loss of neurons in AD is not yet fully understood, a number of theories have been proposed, based largely on the application of molecular genetics. This has led to the discovery of mutations in genes that cause (eg the β-amyloid precursor protein found in the core of senile plaques), or increase the susceptibility to, AD (eg the presence of the proteins presenilin 1, presenilin 2 and apolipoprotein E4), or induce Alzheimer-related pathology (eg the tau protein). This knowledge will provide the foundation from which we develop therapies to slow, or even halt, the disease. Scientists estimate that delaying the mean onset of AD by approximately five years would reduce the numbers of persons with AD by 50 per cent by the year 2050.
Outlook
Since they were first observed by Alzheimer over a century ago, our understanding of the nature of plaques and tangles has increased substantially.6 The prospect of slowing or even stopping their formation now appears to be a realistic possibility. This coupled with approaches to treat the symptoms of AD means that the therapeutic prospects for this debilitating disorder have never been better.
Dr Alan Palmer is chairman of Pharmidex, 72 New Bond Street, London W1S 1RR
The rationale for replacement therapy
Acetylcholine (2-(acetyloxy)-N,N,N-trimethylethanaminium) is an ester of acetic acid (ethanoic acid) and choline. It was the first neurotransmitter to be identified, but was not the first transmitter to be linked to a neurological disorder. This distinction goes to the neurotransmitter dopamine.
In Austria in 1960, two researchers, Herbert Ehringer and Oleh Hornykiewicz, demonstrated that Parkinson's disease is associated with reduced concentrations of dopamine and its major metabolite (homovanillic acid) in a part of the brain called the striatum. This loss was subsequently found to correlate with both cell loss from the substantia nigra (dopamine neurons project from this structure to the striatum) and two of the three cardinal symptoms of Parkinson's disease, ie lack of movement and tremor. (The disease was described as a 'shaking palsy' by the English physician James Parkinson in 1817.) This laid the basis for replacement therapy. Dopamine itself was tried, but it did not cross the blood-brain barrier (BBB), so dopamine's immediate precursor, l-dihydroxyphenylalanine (l-DOPA), which did cross the BBB, was then used instead. l-DOPA is actively transported into the brain by the neutral amino acid transporter and, once peripheral metabolism was blocked, it was used to great effect in the treatment of Parkinson's disease. This ground-breaking work stimulated a number of other groups across the world to begin to investigate the biochemical basis of other neurodegenerative diseases. If there was a loss of dopamine in Parkinson's disease and replacement therapy with l-DOPA provided a rational and effective approach to therapy, then the same logic could be applied to the cholinergic deficit in Alzheimer's disease.
Acknowledgements
My thanks to Steven Palmer for his expert assistance in the preparation of Fig 1.
Related Links
Information on memantine
References
- A. M. Palmer and P. T. Francis in Principles and practice of geriatric medicine, 4th edn, M. S. J. Pathy, A. J. Sinclair and J. E. Morley (eds), pp59-67. Chichester: John Wiley, 2006.
- A. M. Palmer, Neurodegeneration, 1995, 5, 381.
- A. M. Palmer, Investigational Drugs, 2003, 4, 833.
- N. A. Clarke and P. T. Francis, Expert Rev. Neurother., 2005, 5, 671.
- See Memantine website.
- A. M. Palmer, Trends Pharmacol. Sci., 2002, 23, 426.


No comments yet