


Natural products formed the basis of our medicines for most of the 20th century. Although modern synthetic methods promised new drugs in the 1970s, this has not generally proved to be the case. Chemists are once again turning to Nature to replenish the medicine chest.
-
Antibiotics, anticancer drugs, statins and contraceptive steroids - more than 50 per cent of today's drugs are either natural products or have structures based on natural products
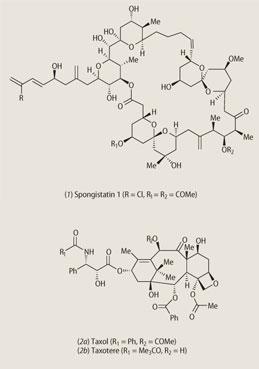
The study of the chemistry of natural products can be dated to the start of the 19th century with the isolation and characterisation (but not structure elucidation) of morphine (1815), quinine (1820) and atropine (1833). These faltering first steps can be contrasted with recent total chemical syntheses of complex marine natural products like the spongistatins (1) (by Steven Ley and his team at the University of Cambridge in 2005), which are remarkable for their elegance and efficiency, and also because they have provided gram quantities of these anticancer agents for pre-clinical evaluation.
Early folk medicine
Without a doubt the primary interest has always been the fascinating and often useful biological properties of natural products. Long before any chemistry was attempted, our ancestors explored the use of plant and animal extracts as adjuncts to hunting, murder and euthanasia; for magical and pseudo-religious purposes; and as medicines or tonics.
The Ebers papyrus of Ancient Egypt (ca1500 BC) has numerous references to the healing properties of plant and animal extracts. Castor oil and beer, for example, are recommended to get rid of the excrement in the body, while the plant lore of the Greek physicians Dioscorides (ca 100 AD) and Galen (129 to 199 AD), provided the basis of prescribing natural medicines for at least the next 1500 years. Henbane and mandrake, and their associated tropane alkaloids, for pain relief is just one example.
A 20th century legacy
The heyday for natural products' chemistry was undoubtedly the first 80 years of the 20th century. During this period huge advances were made in the isolation, purification and structure elucidation of natural products. This was, in the main, made possible by advances in spectroscopic methods - IR, NMR, and mass spectrometry - but also by developments in synthetic methodology which coped better with the great complexity of the structures that were revealed. A greater understanding of how these molecules were assembled (their biosynthesis) was also made possible by the availability of the radioisotopes 3H (tritium) and 14C, and the non-radioactive isotopes 2H (deuterium), 13C and 15N.
The major legacy of these new endeavours has been the incorporation of a large number of natural products into the World's pharmacopoeias. Even today, more than 50 per cent of all drugs are either natural products or have structures based upon natural products - in cancer chemotherapy the percentage is even greater.
A casual examination of a list of the World's most used drugs reveals:
- the statins for lowering blood cholesterol levels (based on the natural products compactin and mevinolin);
- the broad spectrum antibiotic Augmentin - a mixture of the mould product clavulanic acid and the penicillin amoxycillin (based upon the mould product penicillin G); and
- the contraceptive steroids, based on the natural steroids progesterone and estrone.
All of these are produced in vast quantities as are the even better known natural product drugs like morphine, hyoscine and quinine, though most of the latter compound ends up in tonic water!
The apogee of natural product drug research occurred in the period 1950-80. Hundreds of mould-derived antibiotics were isolated and commercialised (penicillins, cephalosporins, aminoglycosides, macrolides), and dozens of plant and mould-derived anticancer agents were discovered and developed, rendering several cancers curable for the first time.
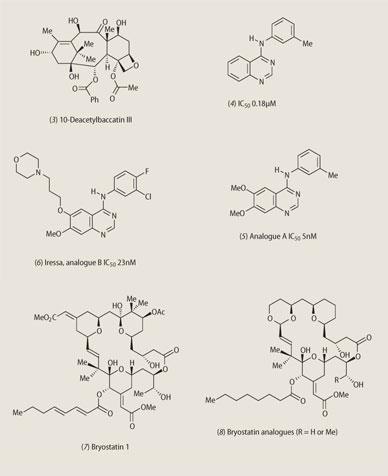
Expeditions were mounted to the wilder places on Earth and even the oceans began to yield new natural product treasures. The pharmaceutical industry and large national research centres like the National Cancer Institute in Washington DC embarked upon screening programmes that examined thousands of natural products each year. Drugs like taxol (2a) (for breast and ovarian cancer) and vancomycin (the antibiotic of 'last resort' for MRSA bacterial infections) were discovered in this way.
The taxol story

The history of the development of taxol (2a) as an anticancer agent is particularly instructive since it has parallels with the discoveries of many other natural product drugs.
The compound, from a crude extract of the bark of the Pacific yew tree, Taxus brevifolia, was first identified as an active anticancer agent in 1971 by Monroe Wall and Mansukh Wani of the Research Triangle Institute in North Carolina. However, the small amounts of compound available (ca 500 mg from 12 kg of bark) meant that pharmacological evaluation was not going to be easy.
In 1979, Susan Horvitz of the Albert Einstein College of Medicine in New York, demonstrated that taxol disrupted cell division (mitosis) by stabilising the microtubules 'that hold the two daughter cells' together during their maturation. This was in direct contrast to the mode of action of the other well known anticancer alkaloids, vinblastine and vincristine, which caused depolymerisation of the microtubules and thus also disrupted cell division.
Early clinical trials with taxol showed that it produced some dramatic responses in patients with breast and ovarian cancer, and a full-scale clinical trial was mounted in the early 1990s. This necessitated the production of up to 2 g of taxol per patient in a trial that envisaged treating 12 000 patients per year.

The Pacific yew might have been eliminated but for a timely discovery made by the groups of Pierre Potier (Paris) and Andrew Greene (Grenoble). They showed that needles of the European yew, Taxusbaccata, contained relatively large amounts of a related natural product 10-deacetylbaccatin III (3).
They and other chemists around the world set about devising chemical routes for the conversion of (3) into taxol. This was a hugely successful project and ensured the supply of both taxol and a French analogue taxotere (2b).
The story is completed by the first total syntheses of taxol by the American groups of Kyriacos Nicolaou and Robert Holton, who reported their respective routes in the same week of February 1994. Both routes are fine examples of modern synthetic chemistry but are too long to be used commercially, so are elegant rather than practical.
Natural products in recess
There were, however, diminishing returns from this vast amount of investigation, and few new natural product drugs were discovered in the 1970s. So the pharmaceutical industry tried a new route for drug discovery that employed combinatorial chemistry coupled with rapid throughput biological screening.
This involved the automated production of tens of thousands of synthetic compounds (libraries of analogues) where the structures had small and systematic alterations. The biological evaluation was also automated by fast throughput screening, whereby compounds are incubated with cells or enzymes in large batches, and the resultant colour changes or cell numbers resulting from biological interactions can be measured automatically.
These two procedures used in tandem allowed companies to examine vast numbers of new structures each year and to construct structure-biological activity profiles. However, disappointingly little has emerged so far from these endeavours, though the design of the anticancer drug Iressa benefited in the early stages of its development from library synthesis. The development of Iressa reveals how the lead structure (4) (50 per cent inhibition of cancer cell growth at a compound concentration of 0.18 μM) led on to more active compounds (5) from the 'library', and to Iressa (6) with good activity and also the best bioavailability.
Overall, the timescale from the discovery of an active compound to its acceptance into the clinic as a potential drug has not decreased significantly from the average of 12-15 years associated with a more conventional route of drug discovery.
Natural products make a comeback
So where does this leave natural product chemistry - the answer is very much back in vogue. Owing to our greater understanding of the three dimensional interactions between natural products and the enzymes and receptors with which they associate, it is now often possible to understand how they exert their biological effects. This means that it is easier to identify the key structural features necessary for optimum binding to a receptor or enzyme active site.
These basic skeleta are called pharmacophores, and they are simpler targets for synthesis than the more complex natural products themselves. A recent example of this new strategy of drug discovery is provided by the bryostatins (7) and (8). The bryostatins are produced by the marine bryozoan Bugula neritina found off the coast of California, and though only isolated in very small quantities, have none the less shown promising anticancer activity.
A total chemical synthesis of these complex structures can take up to 60 chemical steps, so producing these compounds on even the gram scale is potentially a major problem. However, Paul Wender at Stanford University in California has synthesised (in around 20 chemical steps) simplified structures that he believes may represent the pharmacophore of these natural products. These new structures have much more potent anticancer activity and promise to be of considerable interest in the clinic.
A more futuristic route to natural products involves 'combinatorial biosynthesis'. An impressive example is provided by new structures based upon the broad-spectrum antibiotic erythromycin (9). The genes coding for the enzymes that catalyse the various stages of erythromycin biosynthesis have been identified, and through a process of gene shuffling and gene substitution/addition, it is possible to change the sequence of chemical reactions that produce the natural antibiotic.
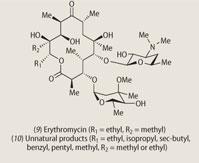
The result is a library of novel structures (10) that can be screened for biological activity, and since these engineered genes can be inserted into microorganisms like E. coli, it should be possible to produce other useful compounds on a large scale by using fermentation-like technology. The next generation of novel natural products are just as likely to be discovered in this way as in the Amazonian jungles or the oceans.
So natural product chemistry is alive and well. The complex structures of natural products have evolved as the genes and encoded enzymes that produce them have evolved. The majority of natural products have some kind of biological activity since they have evolved alongside the enzymes and protein receptors of their host organisms, which have had to compete with other organisms for their ecological niche on the planet.
The structural information for interaction with proteins and enzymes is thus built into their privileged structures which would usually also be the pharmacophore. In many instances the natural products have acted as feeding deterrents or actual toxins, which has enhanced the competitiveness of the producer organisms, and this has ensured the retention of the genes (and enzymes) responsible for their production.
New synthetic analogues will be successful as long as they retain enough of the privileged structures of the parent natural products, which will provide additional drugs for Nature's already well-stocked medicine chest.
Professor John Mann is McClay chair of biological chemistry in the school of chemistry at Queen's University Belfast, Belfast BT9 5AG.
Further Reading
- J. Mann, Murder, magic and medicine, 2nd edn. Oxford: OUP, 2000.
- J. Mann, Life saving drugs. Cambridge: RSC, 2004.
- D. J. Newman, G. M. Cragg and K. M. Snader, Natural Prod. Rep., 2000, 17, 215.


No comments yet