


The first draft sequence of the human genome, announced 10 years ago, was time-consuming and expensive. Now, genome sequencing is fast, simple and affordable, allowing scientists to do what every dividing cell does: to read a single molecule.
-
The first human genome took months to sequence using a technique from 1975
-
Single molecule techniques could potentially do the same in hours
Ten years ago, US president Bill Clinton and UK prime minister Tony Blair announced the draft version of 'the' human genome as a major scientific revolution. In many ways, however, it was a big-budget application of old established technology. The genomics expert and Nobel laureate Sir John Sulston compared it to a moon shot, which may have been both a warning not to expect immediate benefits, and a reflection on the primitive technology used to reach a highly visible goal.
The real revolution has only started to happen. Technology has developed at an immense rate, and costs have fallen dramatically. A recent milestone was the first publication of the personal human genome of Stanford academic Stephen Quake by single molecule technology.
Sequencing technology

The publicly funded human genome sequencing effort, coordinated by Francis Collins in the US and John Sulston in the UK, was based on technology that has been in use since 1975, ie the Sanger sequencing method.
Named after Frederick Sanger, who won separate Nobel prizes for his sequencing work on proteins and on nucleic acids, this method, also known as the chain termination method, relies on copying the DNA in the presence of a defective version of one of the DNA bases, or building blocks, leading to a termination in the building up of a DNA strand.
Applying this principle to four separate samples, with each sample using a different base as the chain terminator, leads to four mixtures of chains of different lengths but (in each vessel) that all end with the same base. If the four chain termination bases are labelled with fluorescent dyes of different colours, the four samples can be in the same reaction vessel to make the process simpler.
In the 1980s, scientists analysed these DNA mixtures by gel electrophoresis, pulling the samples through a gel to give fluorescent bands at different positions, relating to the lengths of the chain fragments. Later, capillary electrophoresis improved the efficiency, but the essential method remained unchanged.
Advancing this technology to sequence his personal genome in a race with the publically funded effort, American biologist John Craig Venter used the same technique, but combined it with his trademark 'shotgun' approach. This approach creates stretches of DNA for sequencing by random fragmentation, and then relies on computer programs to assemble the reads afterwards.
Second generation machines
By 2005, second generation genome sequencing machines were on the market. These no longer required at least one copy (and more likely, thousands of copies) of DNA for every single base to be read. This latter requirement was a drawback of Sanger's method, which made it impossible to simplify it to the point where the information is read out from a single molecule of DNA.
Instead, two of the newer instruments observe directly how a new DNA strand is synthesised, and each new base incorporated is identified via a fluorescence marker ('sequencing by synthesis'). Although the techniques are the same the instruments differ in how the bases are read.
The Illumina Genome Analyser 2 uses the Solexa technique, whereby random fragments of the genome to be sequenced are immobilised in a flow cell, and then amplified in situ, resulting in localised clusters of around 1000 identical copies of each fragment (Fig 1). Sequencing occurs with the help of reversible terminators. These are modified building blocks that stop the DNA synthesis as in the Sanger method, but once a base has been read out, the blocking and the fluorescent label are removed, and the next base can be attached and read.
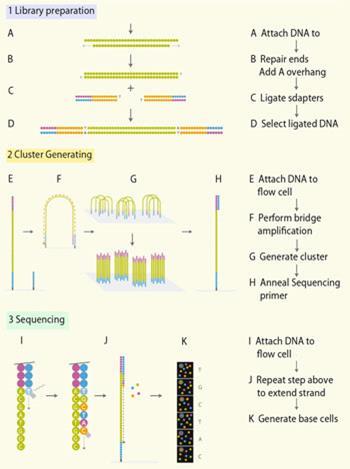
The 454 system, introduced in 2005 and used to sequence the personal genome of DNA pioneer James Watson and that of a nameless Neanderthal, uses pyrosequencing. This technique is based on detecting the pyrophosphate group that is released as a new base is added. (The activated base required for DNA synthesis contains a chain of three phosphate groups, one of which bonds to the DNA to create a phosphodiester linkage and the other two are released as pyrophosphate.)
A third instrument, designed by Applied Biosystems in California, US, uses a mixture of all possible oligonucleotides of a given length and observes them as they are linked together by a ligase enzyme to match the existing strand ('sequencing by ligation').
Each of these three instruments has its strengths and weaknesses in terms of the length of DNA that can be read in one go, and the error rates. If researchers are investigating DNA that is broken into short fragments anyway - for example DNA from fossil specimens - they may value precision over read length. If, by contrast, they need to read highly repetitive sequences - in the human Y chromosome, for instance - the read length will be the crucial parameter.
In the autumn of 2009, these three technologies were busy sequencing around 10 Terabases (1012 bases), equivalent to the information content of 3000 human genomes, per week worldwide (a quarter of that activity was happening at the Sanger centre at Hinxton, near Cambridge). All three still require multiple identical copies of the DNA, so small samples have to be amplified using the polymerase chain reaction (PCR). In PCR, the DNA double helix is repeatedly separated into its two complementary strands by heating, allowing a heat-stable enzyme such as Taq polymerase to build a new daughter strand that is complementary to each of the original ones. This cycle can then be repeated indefinitely doubling the copy number every time like a chain reaction. Therefore, PCR can produce a large amount of a specific DNA strand even when starting from tiny amounts.
All of these instruments also rely on fluorescence detection, which requires bulky and expensive optical equipment that needs to become even larger when the sample size is reduced.
The next generation
Ideally, in the next generation sequencing technology, both the PCR and the bulky optical equipment would disappear. Such instruments would be able to read a single molecule of DNA, detecting each base by an electronic signal rather than an optical one. Scaling down to single molecules would reduce the cost of chemicals to negligible amounts, while the switch from fluorescence to electronics would make the equipment much simpler and more affordable.
Several companies are working on these goals. Indeed, Helicos Biosciences, based at Cambridge, Massachusetts, US, already has a single molecule sequencer on the market. This 'HeliScope' technology is based on sequencing by synthesising DNA in a flow cell using fluorescence detection with a microscope. Essentially, one kind of fluorescence-labelled base is added at a time, then the location of the label is imaged before the label is cleaved off and washed away, so another base can be added. For each base to be sequenced, all four bases have to be tried out, with only the one successful incorporation showing up on the image taken (Fig 2). This method was used in the sequencing of the personal genome of Helicos co-founder and Stanford (US) academic Stephen Quake, published in August 2009.1

Possibly the next contender to reach the market place will be the one designed by Pacific Biosciences. This instrument also uses a fluorescence-based sequencing-by-synthesis approach. The 'trick' that allowed the company to reach single molecule readout is its reaction chamber, which is so small that only one fluorescent molecule at a time can ever be in it, eliminating background fluorescence, or noise.
Moreover, the scientists at Pacific Biosciences in California, US, attach the fluorescent label to the phosphate group of the base to be incorporated, which ensures that the label is cleaved off as the phosphodiester bond is formed to attach the base to the growing chain. In the traditional technology, the label would be attached to the ribose and thus end up in the synthesised DNA strand, contributing to unwanted fluorescence background.
Oxford University spinout Oxford Nanopore Technologies is developing a radically different approach to single molecule sequencing, in which no fluorescent label is needed, and the bases are read out one by one as they are cleaved off the DNA strand ('sequencing by degradation'). The approach is based on the research of Hagan Bayley at Oxford University, who investigated the properties of the α-hemolysin membrane pore, a protein which the pathogen Staphylococcus aureus produces in order to punch holes into the cell membranes of its hosts.
Oxford Nanopore uses a modified version of this natural membrane pore to record the passing of each individual DNA base through the pore after it has been cleaved off the DNA strand being sequenced (Fig 3).
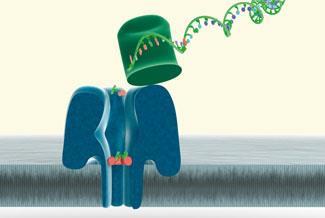
The pore connects chambers with different ion concentrations, so that under normal circumstances an ion current flows through the pore and an electrical signal can be measured. DNA bases cleaved off from the DNA strand by an enzyme (an endonuclease) migrate through the pore slowly (inhibited by a specifically designed bottleneck, based on cyclodextrin chemistry), thus blocking the current for a characteristic length of time. This detection method yields different electronic signals for the four standard bases and for methylated cysteine.
In a paper published in February 2009, researchers at Oxford showed that the modified pore can not only distinguish between the four standard bases, but can also recognise methylated cysteine, which is important for the study of epigenetics - the study of heritable traits (over rounds of cell division) that do not change the underlying DNA sequence, for example post-translational modifications like methylation.2 What the company still has to optimise is the attachment of the DNA-digesting enzyme near the entrance to the pore, to ensure that every base cleaved off passes through the pore and is counted.
Future applications
When the single-molecule, fully electronic genome analysers reach the market, they are likely to revolutionise medical research and diagnosis. Cancer - its prevention, diagnosis, and treatment - is one of the most urgent targets for genome research. Following up on an earlier sequencing of leukaemia cells,3 six genome studies of cancer patients and/or cell lines appeared in scientific journals by January 2010, demonstrating a large number of changes in the genomes of cancerous cells, compared to the genomes of healthy cells from the same patient.
To understand which genetic alterations have caused or benefited the propagation of a cancer, researchers will need hundreds of examples of such genome pairs for each type of cancer.
With the next generation of genome sequencers, this amount of genomic information will become accessible. Going one step further, sequencing experts are also expecting that doctors may soon be able to sequence the genome of an individual patient's tumour cells to find out which drug will be most suitable to treat the disease and the dosage that the patient would need.
As scientific journals celebrate the 10th 'birthday' of the human genome, it becomes clear that the genome revolution that we were promised in 2000 hasn't really happened yet. But with the current work from groups around the globe it's imminent.
Michael Gross is a science writer based at Oxford. He can be contacted via his web page.
References
- D. Pushkarev et al, Nature Biotech., 2009, 27, 777.
- J. Clarke et al, Nature Nanotechnol., 2009, 4, 265.
- T. J. Ley et al, Nature (London), 2008, 456, 66.


No comments yet