


Every year between one and two million people - mainly children - living in the tropics and subtropics die of malaria.
Every year between one and two million people - mainly children - living in the tropics and subtropics die of malaria. Artemisinin is the most effective treatment against malaria, the most infectious disease in the world today. What hope does this drug offer for the future?

Malaria is caused by the bite of a female mosquito (Anopheles), resulting in the malaria parasite, Plasmodium, entering the human blood stream. Once a red blood cell has become invaded with the parasite, several rounds of asexual reproduction ensue, leading to its eventual rupture. It is the cyclic release of parasites from the red blood cells which causes the intermittent symptoms of fever, shivering and anaemia that are characteristic of malaria.
Of the four species of Plasmodium that infect humans, the most dangerous is P. falciparum. This parasite accumulates in the capillaries of vital organs such as the brain, kidney, intestine and lungs. Cerebral malaria, the accumulation of P. falciparum in the brain, is responsible for most of the deaths associated with malaria.
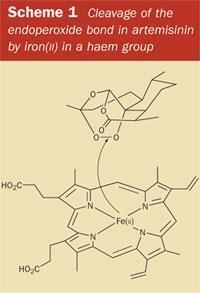
During the red-cell cycle, the parasite uses the host's haemoglobin as food. The haemoglobin protein is broken down by enzymes and the parasite assimilates the amino acids which are released. The digestive process also liberates haem, the iron-containing porphyrin which is normally buried within the haemoglobin molecule. It is this free haem molecule, bearing an exposed Fe(II) atom at its centre, which provides the unique line of attack for the drug artemisinin (1).
The discovery of artemisinin
The story of artemisinin began in China in the late 1960s. After a brief respite, lasting only a few decades, malaria was once again on the increase. DDT, at first heralded as the insecticide that would eradicate the mosquito, was being withdrawn because the vector had developed resistance to it. Chloroquine (2), developed by the US military towards the end of World War II to kill the Plasmodium parasite, was also losing its effectiveness in much of South East Asia, South America and - increasingly - in Africa.1 (Chloroquine inhibits the polymerisation of the toxic haem group, an accumulation of which kills the parasite.) In the absence of an effective insecticide or an effective vaccine, a new antimalarial drug seemed to be the only hope.

Against this scene of increasing resistance, and of the on-going wars in neighbouring Cambodia and Vietnam, the Chinese Government began a systematic examination of plants used in traditional Chinese medicine with the aim of finding a new antimalarial. The herb Artemisia annua, or 'qinghao', was one of several hundred species listed in the Chinese pharmacopeia to be considered. According to its listing, A. annua is cool in nature (yin) and can therefore be used to treat internal heat conditions (yang). In fact, the first report of the use of qinghao in this context had appeared in 1596 AD - in the Chinese materia medica, Bencao Gangmu - for treating 'hot and cold due to intermittent fever illness'. According to the old pharmacopeias, one bunch of the leaves, collected in spring or summer, was taken with two 'sheng' of water and pounded with a pestle and mortar to express the 'juice'. (This procedure was, perhaps, intended to improve the recovery of essential oils from the leaves, in which the active principal, artemisinin, as it is called outside China, is now known to be most concentrated.)
However, when the Chinese scientists made hot water extracts of A. annua according to the ancient texts, they observed no activity against mice infected with the rodent malarial parasite, Plasmodium berghei. Fortunately though, they also tested cold ethereal extracts of qinghao. These did show encouraging activity, and it was not long before 'qinghaosu' (meaning 'principle from qinghao' in Chinese) - a stable and easily crystallisable compound - had been isolated and characterised.
In the 1970s clinical studies with the extract on members of the Chinese army infected with the Plasmodium parasite demonstrated an outstanding therapeutic efficiency in almost all patients treated. Even more impressively, there were no obvious side effects (though rumours of an associated neurotoxicity did appear at the time). Most importantly, qinghaosu was completely effective in treating chloroquine-resistant falciparum malaria, and its often fatal complication, cerebral malaria.
However, even though the Chinese scientists had demonstrated that qinghaosu could clear the blood of malarial parasites more quickly than any other drug, it was a decade before artemisinin would make its appearance on the global stage. The cold war dragged on, and communist China remained suspicious of the West. International organisations, such as the World Health Organisation (WHO), which had the resources to develop new antimalarials, were denied access to both the drug and the herb. Furthermore, some Western scientists were sceptical about the 1,2,4-trioxane ring embedded in the structure of artemisinin. They doubted whether such a functional group could be stable and questioned whether artemisinin could ever really be useful as a drug.
First-generation drugs derived from artemisinin
We now know that the unusual structural feature of artemisinin, the 1,2,4-trioxane ring, is the basis for the unique antimalarial action of the drug. It is the endoperoxide linkage in the ring which 'triggers' artemisinin to 'explode' - but only in the vicinity of the Plasmodium parasite. The endoperoxide bond is cleaved when it comes into contact with iron(II), releasing reactive radicals which ultimately destroy the parasite (see Scheme 1). Substantial quantities of reactive iron(II) accumulate inside an infected red blood cell as a result of the liberation of the haem group, which is a byproduct of the digestion of haemoglobin. However, the effective 'delivery' of the artemisinin 'bomb' to the infected red blood cells where it can be detonated, suffers from one fundamental problem, which the Chinese scientists had already encountered when attempting to prepare their herbal teas from qinghao. Artemisinin is poorly soluble in water (and also in oil). Therefore, when creating the first generation of drugs derived from artemisinin, the overriding goal was to improve its solubility characteristics, so that the new drug might be more easily formulated and more efficiently delivered.
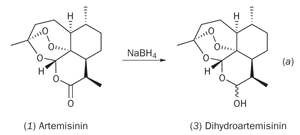
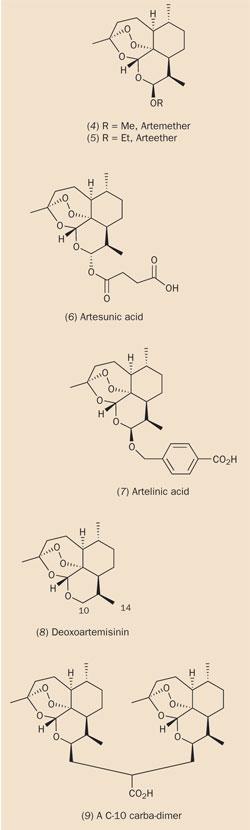
The most accessible functionality in artemisinin is the lactone group, so it was here that chemists made the first chemical modifications to the natural product. Reduction of artemisinin by sodium borohydride (see equation (a)) produced dihydroartemisinin (3), in which the lactone group had been converted to a lactol (hemi-acetal) group. The intact 1,2,4-trioxane ring in dihydroartemisinin retained a potency greater than that of artemisinin itself, though dihydroartemisinin lacked stability in vivo. However, this problem could be solved by attaching a substituent to the free hydroxyl group. In addition, by making a careful selection of this substituent, it was also possible to control the solubility of the resultant drug. Several hundred such derivatives were prepared in the 1980s and early 1990s, incorporating substituents attached by ether, ester or carbonate linkages. Structures (4)-(7) represent a few of the more successful semi-synthetic first generation drugs for the treatment of malaria.
The ether-substituted drugs, artemether (4) and arteether (5), are both soluble in oil (they can be formulated in sesame oil or peanut oil), and can be administered by intramuscular injection. Both compounds are metabolised quite rapidly to dihydroartemisinin in vivo. However, the treatment of advanced cases of malaria requires a water-soluble analogue of artemisinin, which can be injected intravenously, and thus delivered more quickly than by the intramuscular route. Artesunic acid (6), the succinate ester of dihydroartemisinin, is one such derivative. The only drawback is that the hemi-ester linkage in artesunate is unstable, and this derivative is hydrolysed to dihydroartemisinin even more rapidly than the ethers. Artelinic acid (7), the p -carboxybenzyl ether of dihydroartemisinin, which was developed by the Walter Reed Army Institute (US) as an orally-effective antimalarial, retains the favourable characteristics of water-solubility found in artesunate while overcoming its hydrolytic instability. Current malaria therapy is based on both artemether and artesunate.
Second- and third-generation antimalarials
After the success of the first generation of artemisinin-derived drugs, researchers have focused on designing more robust analogues of artemisinin with increased metabolic stability. They have found that the lactone carbonyl group in artemisinin can be removed completely without any detriment to activity. In the latter half of the 1990s, a series of such C-10 carba-analogues were synthesised, (8)-(11), based on deoxoartemisinin (8), which is eight-fold more active than artemisinin in vitro. (C-14 derivatives were also found to be effective.) Of particular interest was the discovery that some C-10 carba-dimers, eg (9), were not only potent antimalarials, but also demonstrated anticancer properties in vitro.
The effect of including substituents at other positions in the artemisinin system has also been extensively investigated. One analogue, incorporating a methyl group at the 3-position, proved particularly effective, perhaps because this substituent enhanced the stability of a (putative) primary radical which was generated by endoperoxide cleavage (see Scheme 1). Conversely, chemists have found that introducing substituents on the α-face of the artemisinin molecule should be avoided. This supports the 'triggering' mechanism because the presence of an α-substituent would disrupt the tight binding between artemisinin and haem, which is proposed to activate the drug. The most extensive manipulations of the artemisinin 'lead compound' have revealed that even when the B and D rings were removed completely (as in compounds (10) and (11)), the resultant molecule still retained antimalarial activity.
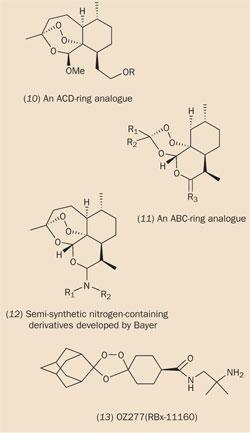
Major pharmaceutical companies are, for the first time in recent history, beginning to take an interest in developing a new generation of antimalarial drugs derived from artemisinin. The German pharmaceutical company Bayer has synthesised and screened several semi-synthetic nitrogen-containing derivatives, eg (12), in collaboration with Richard Haynes of the Hong Kong University of Science and Technology.2 One of these compounds, artemisone (structure not available), is currently undergoing phase II clinical trials.
Jonathan Vennerstrom of the University of Nebraska, with support from the Medicines for Malaria Venture (MMV) and Hoffman-La Roche, has been designing a new generation of completely synthetic drugs, which are inspired by artemisinin, but not derived from it. These drugs, for example OZ277 (13), retain the 1,2,4-trioxane ring found in the lead compound.3 When designing this compound, the main criteria were to achieve low production costs (ie via a straightforward synthesis and simple formulation) together with a maximum of a three-day treatment regime, based on a once-daily administration (ie good potency and pharmokinetic properties and a low potential for toxicity).
The synthesis of OZ277 ought to be inexpensive, because it is an ozonide. Ozonolysis involves the cleavage of a carbon-carbon double bond, leading to two products at the aldehyde/ketone level of oxidation. However, the reaction actually proceeds via an ozonide intermediate, in which three oxygen atoms are constrained in a five-membered ring. These ozonides can often be isolated if the work-up is not done under reducing conditions. The ozonide ring in OZ277 is substituted on one side by a stable adamantane group, and on the other by a cyclohexyl ring, incorporating both an amide and a primary amine functional group. It is this second half of the molecule which provides the 'fine tuning' that allows it to be used as a drug. By making a judicious choice of this moiety, it has been possible to retain a nanomolar antimalarial activity, without compromising either the in vivo stability of the ozonide or its aqueous solubility, all of which are necessary for the compound to be formulated as a pill. The first clinical trials of OZ277 took place in Thailand earlier this year.
The future of artemisinin
Despite much research, artemisinin remains the only known natural product to contain a 1,2,4-trioxane ring, and Artemisia annua continues to be the only known natural source. (Phytochemical investigation of this species has revealed an abnormally wide range of other endoperoxides and hydroperoxides, many of which have not, as yet, been tested for their antimalarial activity).4 Several total syntheses of artemisinin are reported but, because of the complexity of the target compound, none is commercially competitive with extraction from the natural source. In addition, although micropropagation (tissue culture) of A. annua can be easily accomplished, it is equally unlikely that in vitro production of artemisinin will become a commercially viable proposition. It seems that the production of artemisinin from wild or field-cultivated plants will be the preferred option for the foreseeable future.
This method of production is currently practised, on a multi-tonne scale, from plants growing in the wild in China and Vietnam. Many Chinese scientists believe that, because of its low price (just a few hundred US dollars per kilogram), artemisinin will continue to be a cheap and readily available starting material for the synthesis of improved analogues. However, the semi-synthetic drugs which are derived from artemisinin can sometimes be several orders of magnitude more expensive than the source material itself. This has prompted some US scientists to claim that semi-synthetic artemisinin derivatives are too expensive for the developing World, where they are needed most. They argue that this cost issue therefore justifies the investigation of alternatives, which are independent of the natural source.

Artemisinin is currently at the heart of the WHO's global fight against malaria, and a new generation of drugs, based on artemisinin, should reach the market in a few years time. Remarkably, there has still not been one documented clinical case of resistance to artemisinin or any of its derivatives. But how long can this situation last? A report published last year5 suggests that resistance may now be arising in areas with uncontrolled use of artemisinin derivatives. The researchers concluded that this unexpected sudden rise in resistance indicates the need for increased vigilance and a coordinated and rapid deployment of drug combinations. Indeed, when treating both multidrug-resistant and non-drug resistant falciparum malaria, it is now recommended by WHO that artemisinin be taken in combination with another drug (this is a strategy designed to slow the development of resistance, since during treatment with two - or more - drugs, the chance of a mutant emerging which is resistant to both is drastically reduced). In the continuing absence of an effective malaria vaccine, the development of new antimalarial drugs - most likely derived from, or inspired by, artemisinin - will continue to be our primary weapon in the fight against malaria.
Dr Geoff Brown is a lecturer at the school of chemistry at the University of Reading, Whiteknights, PO Box 217, Reading RG6 6AH.
References
- A. Butler and T. Hensman, Educ. Chem., 2000, 37 (6), 151.
- R. K. Haynes et al, Angew. Chem. Int. Edn, 2004, 43, 1381.
- J. L. Vennerstrom et al, Nature (London), 2004, 430, 900.
- G. D. Brown, G.-Y. Liang and L.-K. Sy, Phytochemistry, 2003, 64, 303.
- R. Jambou et al, Lancet, 2005, 366, 1960.






No comments yet