


Trifluoromethyl sulfur pentafluoride - a byproduct of the electronics industry - has been named a 'super' greenhouse gas by physical chemists. But what evidence do they have that makes this molecule a potential threat to the environment?
-
Data from ice reveal CF3 SF5 concentration in atmosphere is rapidly rising
-
CF3 SF5 has an exceptionally long lifetime in the atmosphere, a further cause for concern
Over the past seven years the importance of greenhouse gases and global warming has leapt from obscurity to the top of the political agenda in all developed countries, culminating in the Stern report in November 2006 when the economic consequences of unchecked global warming were spelt out. Here we review the science of the greenhouse effect - or, more accurately, radiation trapping - and describe what constitutes a serious greenhouse gas, taking CF3SF5 as a case study.
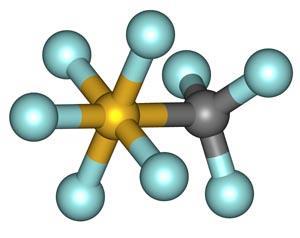
CF3SF5 in the atmosphere
CF3SF5 is a gas at room temperature with a boiling point of 253 K and an enthalpy of vaporisation of 20 kJ mol-1. The gas was first detected in the Earth's atmosphere in 2000,1 by which time the related SF6 molecule had already been identified as a potential greenhouse gas.2 The source of CF3SF5 is believed to be anthropogenic, and most likely a breakdown product of the dielectric molecule SF6 in high-voltage equipment. The trends in concentration levels of SF6 and CF3SF5 have tracked each other closely over the past 30-40 years (Fig 1 (a)), suggesting that CF3SF5 is probably produced in the electronics industry via the recombination of CF3 and SF5 free radicals.
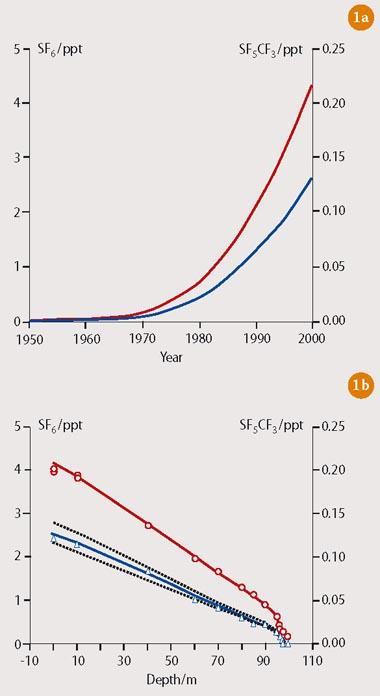
Moreover, measurements taken from ice samples in Antarctica show similar variations of concentration of SF6 and CF3SF5 at depths below the Earth's surface (Fig 1 (b)). Specifically, the data reveal that the concentration of CF3SF5 has grown from near zero in the late 1960s to ca 0.12 pptv in 1999 (or ca 2.5 × 106 molecules cm-3) with a current growth rate of ca 6 per cent per annum. Stratospheric profiles suggest that the lifetime of CF3SF5 in the atmosphere is between several hundred and a few thousand years. So is CF3SF5 a potentially serious greenhouse gas?
The greenhouse effect
The greenhouse effect describes the trapping of infrared radiation, emitted by the Earth, which is absorbed in the atmosphere by small polyatomic molecules such as CO2, CH4 and H2O. As a result the average temperature of the Earth is raised from 256 K, or -17°C, to a hospitable ~290 K. Often called the 'natural' greenhouse effect, this kept the Earth's temperature approximately static for hundreds of years up to the start of the Industrial Revolution, ca 1750.
What we are concerned with today is an 'enhanced' greenhouse effect, caused by increases in concentration of greenhouse gases over the past 250 years. Nobody really doubts the scientific evidence that the concentration of the principal greenhouse gas, CO2, has increased by ca 35 per cent over this period, from ~280 to ~380 parts per million by volume (ppmv), while the average temperature has also increased by ~1°C. What has not yet been proven is that there is a cause-and-effect correlation between these two facts. The consensus of world scientists, and certainly physical scientists,3 is that the CO2 concentration and the temperature of the planet are strongly correlated, but there remains a small vociferous minority who believe otherwise.
Although most attention has been given to CO2 (and possibly CH4 and H2O), physical scientists now understand that there are larger polyatomic gases of low concentrations in the atmosphere which can contribute significantly to global warming. These gases, which include SF6 and CF3SF5 ,1,2 absorb infrared radiation strongly in regions where CO2, CH4 and H2O do not absorb.
Properties of greenhouse gases
In qualitative terms, there are two properties that are necessary for a molecule to be an effective greenhouse gas.
- The molecule must absorb infrared radiation strongly in the black-body range of the Earth's emission, ca 5-25 μm, where CO2, CH4 etc do not absorb. In practice, many C-F and C-Cl stretching vibrations around 10 μm contribute strongly. Such transitions are only observed if the vibration causes a change in dipole moment of the molecule. Figure 2 shows the calculated infrared absorption spectrum of CF3 SF5. The molecule has 24 vibrational modes, but only six have any significant infrared intensity. The four most intense bands occur in the atmospheric window 8-14 μm or 730-1260 cm-1, where the major greenhouse gases CO2, CH4 and H2O do not absorb. (Note that the vibrations of N2 and O2, comprising 99 per cent of the Earth's atmosphere, are infrared inactive.)
- The molecule must have a long lifetime (at least 10 years) in the atmosphere; it must not be destroyed by photodissociation in the range 200-600 nm, and it must not react with the free radicals prevalent in the atmosphere.
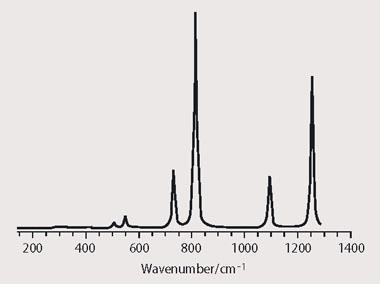
Furthermore, a greenhouse gas whose concentration is increasing rapidly owing to human activity will cause special concern. The gas CF3SF5 satisfies these criteria.
Table 1 shows data for four greenhouse gases - CO2 and CH4 together cause ~70 per cent of the total radiation trapping; a chlorofluorocarbon CF2Cl2; and CF3SF5. The 'radiative forcing' is a measure of the strength of the infrared absorption bands over the range 5-25 μm, it is a per molecule microscopic property with units of W m-2 per unit concentration. When multiplied by the change in concentration of pollutant over a defined time, usually 250 years from the start of the Industrial Revolution to the present day, the macroscopic radiative forcing in units of W m-2 is obtained. We can then compare the effect of different pollutant molecules over a specific period. The greenhouse potential (GHP), sometimes called the global warming potential, measures the radiative forcing, Ax of pulse emission of a greenhouse gas, x, over a defined time, t, usually 100 years, relative to the time-integrated radiative forcing of a pulse emission of an equal mass of CO2 (see equation (i)).
The GHP is a dimensionless number that tells us how important one unit of mass (eg 1 kg) of a pollutant x is to the greenhouse effect compared with the same unit of mass of CO2. The GHP of CO2 is defined to be unity. Equation (i) can be simplified to give an analytical expression for the GHP of x over a time, t (see equation (ii)).4
The GHP of x reflects values for the microscopic radiative forcing, ao, of greenhouse gases x and CO2; the molecular weights of x and CO2; the lifetime of x in the atmosphere τx; and the constant K CO2 which can be determined for any value of t. K CO2 has units of time, and is related (but not equal) to the lifetime of CO2 in the atmosphere, the values of which range from 50 to 200 years.4
The macroscopic overall contribution of a pollutant to the greenhouse effect involves a complicated convolution of its concentration, lifetime and GHP value. Thus CO2 and CH4 contribute most to the greenhouse effect because of their high atmospheric concentration (note that the microscopic radiative forcing and GHP of both gases are relatively low). By contrast, CF3 SF5 has the highest microscopic radiative forcing of any known greenhouse gas (earning it the title 'super' greenhouse gas). (Absolute infrared absorption measurements have shown the microscopic radiative forcing of CF3SF5 to be 0.60 ± 0.03 W m-2 ppbv-1, which is even higher than that of SF6.) The GHP of these two molecules is therefore very high - SF6 is slightly higher because its atmospheric lifetime, 3200 years,4 is about three times greater than that of CF3SF5. However, the contribution of these two molecules to the overall greenhouse effect is still relatively small because their atmospheric concentrations, despite rising rapidly, are still low, at the level of parts per trillion by volume.
Lifetime studies
The reactions that remove CF3SF5 from the atmosphere are important because they contribute to the molecule's lifetime and GHP value. The total removal rate per unit volume per unit time is given by equation (iii), where:
-
each of the five terms in the large bracket of equation (iii) is a pseudo-first-order rate constant;
-
[x] represents the concentration of species x, in this case •OH, O•, cations and electrons;
-
the first four terms represent reactions of CF3SF5 with •OH, O•, cations and electrons, respectively;
-
ki are the corresponding second-order bimolecular rate coefficients.
OH radicals and electronically-excited O atoms, O•, are the most important oxidising free radicals in the atmosphere. The first term in the large bracket dominates in the troposphere (0 <altitude (h)<10km) ; the second term in the stratosphere (10 <h <50 km); and the third and fourth terms in the mesosphere (h >50 km). In the fifth term, σλ and Jλ are the absorption cross section for CF3 SF5 and the solar flux, respectively, and Φλ is the quantum yield for dissociation at wavelength λ. In the troposphere, the summation for λ is over the range ca 290-700 nm, in the stratosphere ca 200-290 nm, and in the mesosphere the solar flux at 121.6 nm dominates all other vacuum ultraviolet (vuv) wavelengths. Equation (iii) assumes that the ion-molecule and electron attachment reactions lead to the removal of CF3SF5 by formation of dissociation products. Furthermore, secondary reactions of such products must not recycle CF3SF5.
Experimental results
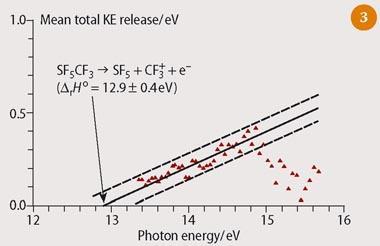
We measured, albeit in an indirect manner, the strength of the CF3-SF5 bond that connects the two radicals.5 We used tunable vuv radiation from the UK Daresbury synchrotron source to photoionise CF3SF5. We measured the translational kinetic energy released as CF3SF5+ dissociated into CF3+ + SF5 as a function of photon energy, and obtained the first dissociative ionisation energy of this molecule, ie Δr H0 at 0 K for the reaction CF3SF5 → CF3+ + SF5 + e- (see Fig 3). The error in each value of the kinetic energy is ca 20 per cent. A linear extrapolation to zero kinetic energy gave the first dissociative ionisation energy of CF3SF5 to be 12.9 ± 0.4 eV. We then determined the strength of the CF3-SF5 bond to be 3.86 ± 0.45 eV or 372 x 43 kJ mol-1, and the enthalpy of formation of CF3SF5 at 0 K to be -1750 ± 47 kJ mol-1. The high dissociation energy indicated a strong σ-bond, a slightly surprising result. This result confirmed, however, that uv photolysis in the stratosphere was unlikely to contribute to the rate of removal of CF3SF5 from the atmosphere.
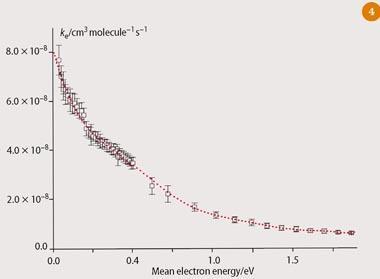
We have since made laboratory-based measurements relevant to the mesosphere, where ionic processes involving cations, anions, electrons and vuv photoexcitation at 121.6 nm dominate. We measured rate coefficients and product yields of small cations reacting with CF3 SF5 in a flow tube, the rate coefficient of low-energy electrons reacting with CF3 SF5, and the absorption cross section of CF3SF5 at 121.6 nm.4 Our results suggest that the dominant process that removes CF3SF5 from the mesosphere is low-energy electron attachment (ca 99 per cent), with vuv photodissociation only making a minor contribution (ca 1 per cent), see Fig 4. (The rate coefficient for electron attachment is measured as a function of mean electron energy, ε, in atmospheric pressure of N2 (ε<0.5 eV) and Ar (ε>0.5 eV). The thermal rate coefficient at 298 K, where ε= 0.038 eV, is 7.7 ± 0.6 × 10-8 cm3 molecule-1 s-1, and the sole product is dissociative, SF5.-6 Ion-molecule reactions make negligible contribution, not because the rate coefficients are too low but because the concentrations of the relevant ions in the mesosphere (eg N+, N2+) in equation (iii) are too small.
Physical chemistry data
To a physical chemist, the lifetime of a greenhouse gas is determined by the inverse of the pseudo first-order rate constant of the dominant chemical or photolytic process that removes the pollutant from the atmosphere.
Consider, for example, CH4. This gas is removed in the troposphere via oxidation by the OH free radical:
•OH + CH4 H2O + CH3
The rate coefficient for this reaction at 298 K is 6.4 x 10-15 cm3 molecule-1 s-1, so the lifetime is approximately equal to (k298 [•OH])-1. Assuming the tropospheric •OH concentration to be 0.1 pptv or 106 molecules cm-3, the lifetime of CH4 is calculated to be ca five years. This is within a factor of 2.4 of the accepted value of 12 years (Table 1). The difference arises because CH4 is not emitted uniformly from the Earth's surface, a finite time is needed to transport CH4 via convection and diffusion into the troposphere, and oxidation occurs at different altitudes in the troposphere where the •OH concentration varies from its average value of 106 molecules cm-3.
Table 1 Examples of four greenhouse gases and their contribution to global warming
| Greenhouse gas | CO2 | CH4 | CF2CL2 | CF3SF5 |
| Concentration (c)/ ppmv | 380 | 1.75 | 0.0005 | 1.2 x 10-7 |
| Change in c (1750-2000)/ ppmv | 100 | 1.05 | 0.0005 | 1.2 x 10-7 |
| Change in c currently/ per cent per year | 0.45 | 0.60 | ca 5.0 | ca 6.3 |
| Microscopic radiative forcing/ W m-2 ppbv-1 | 1.68 x 10-5 | 4.59 x 10-4 | 0.32 | 0.60 |
| Total raidiative forcinga/ W m-2 | 1.46 | 0.48 | 0.16 | 7.2 x 10-5 |
| Lifetimeb/ years | 50-200c | 12 | 100 | ~1000 |
| GHP (100-year projection) | 1 | 23 | 10600 | ~18000 |
| Contribution to GH effect/ per cent | 52 | 17 | ~6d | 0.003 |
|
a Owing to change in concentration of greenhouse gas from the pre-industrial era to the present time; b assumes a single-exponential decay for removal of greenhouse gas in the atmosphere;3c CO2 does not show a single-exponential decay;4d cumulated effect of all chlorofluorocarbons is estimated to be 15 per cent. |
We can regard this as an example of a two-step kinetic process, A → B → C, with first-order rate constants k1 and k2. The first step, A → B, represents the transport of the pollutant into the atmosphere, while the second step, B → C, represents the chemical or photolytic process (eg reaction with an •OH radical in the troposphere, electron attachment in the mesosphere etc) that removes the pollutant from the atmosphere. In general, the overall rate of the process (whose inverse is the lifetime) will be a function of both k1 and k2, but its value will be dominated by the slower of the two steps. Thus, in writing the lifetime of CH4 as (k298 [•OH])-1, we assume that the first step, transport into the region of the atmosphere where chemical reactions occurs, is very fast with k1 k2.
CF3SF5 behaves in the opposite sense, and now k1≪k2. The slow, rate-determining process is the first step, ie transport of the greenhouse gas from the surface of the Earth into the mesosphere, and the chemical or photolytic processes that remove CF3SF5 in the mesosphere will have little influence on the lifetime. We can define a chemical lifetime, τchemical, as (ke [e-] + σ121.6 J121.6 Φ121.6)-1, the value of which will vary with altitude. In the troposphere, τchemical will be infinite because both the concentration of electrons and J121.6 are effectively zero, but in the mesosphere τchemical will be much less. Assuming the electron attachment step is dominant, multiplication of ke for CF3SF5 by a typical electron density in the mesosphere yields a chemical lifetime which is far too small and bears no relation to the true atmospheric lifetime, because most of the CF3SF5 does not reside in the mesosphere. Using calibration data for SF6, the globally-averaged lifetime of ~1000 years for CF3SF5 (Table 2) comes from a weighted integration of the removal rates in the different regions of the atmosphere. Its lifetime is therefore determined by the meteorology that transports it into the mesosphere, and the chemical fate of each molecule when it reacts in that region with low-energy electrons; and radiation at 121.6 nm makes negligible contribution to the atmospheric lifetime.
Table 2 Thermal electron attachment rate coefficients, absorption cross-sections at 121.6nm, and atmospheric lifetimes for CF4, SF6 and CF3SF5. From these data CF3SF5 appears to behave as a perturbed SF6, and not as a perturbed CF4 molecule
| Perfluoro compound | ke (298k)/ cm3 s-1 | σ121.6/cm2 | lifetime/ years |
| CF4 | <10-16 | -22 | >50000 |
| SF5 CF3 | 7.7 x 10-8 | 1.3 x 10-17 | ~1000 |
| SF6 | 2.38 x 10-7 | 1.76 x 10-18 | ~3200 |
Generic lessons
Currently, there are no known undesirable chemical effects of low concentrations of CF3SF5 (and SF6) in the atmosphere. However, their rapidly increasing concentrations and their exceptionally long lifetimes means that life on Earth may not be able to adapt to any changes these gases may cause in the future.
It is worth remembering that the concentration of chlorofluorocarbons in the stratosphere grew from industrially-produced benign molecules to serious ozone-depleting molecules over a period of less than 20 years. Atmospheric scientists are thus investigating the photochemical properties of CF3SF5, which two years ago contributed just 0.003 per cent to the total radiation trapping, reasoning that small problems in this area have a tendency to become big problems.4 According to Professor Ravishankara,7 a physical chemist at the University of Colorado at Boulder, all such exceptionally long-lived molecules should be considered guilty unless proven otherwise.
One solution would be to discover inert, dielectric gases with low GHP values which could be used as substitutes for SF6 in industrial applications. Ring-based perfluorocarbons, such as cyclic-C4F8 and cyclic-C5F8 are possibilities. However, the simplest solution is that we should not put up into the atmosphere any more pollutants than are absolutely necessary.
Richard Tuckett is professor of chemical physics in the school of chemistry at the University of Birmingham, Edgbaston, Birmingham B15 2TT.
References
- C. Fan et al, Proc. Natl. Acad. Sci. U.S.A., 2003, 100, 9134.
- A. A. Lubin et al, Anal. Chem., 2006, 78, 5671.
- Y. Xiao et al, Angew. Chem. Int. Edn, 2005, 44, 5456.
- A.-E. Radi et al, J. Am. Chem. Soc., 2006, 128, 117.
- Y. Xiao et al, J. Am. Chem. Soc., 2005, 127, 17990.
- Y. Xiao et al, Proc. Natl. Acad. Sci. U.SA., 2006, 103, 16677.
- B. R. Baker et al, J. Am. Chem. Soc., 2006, 128, 3138.
- R. Y. Lai et al, Anal. Chem., 2007, 79, 229.
- Y. Xiao et al, J. Am. Chem. Soc., 2007, 129, 262.





No comments yet