


Thirty three per cent of all cancers diagnosed are skin cancers, and the incidence of skin cancer especially among young adults is on the increase. Current treatments using anticancer agents suffer unpleasant side effects and are not particularly efficient. Now, prodrugs - selective chemical agents - are beginning to show potential as a cure for this severe form of cancer. Prodrugs - selective chemical agents - are beginning to show potential as a cure for skin cancer
-
Excessive exposure to uv light initiates melanoma in the skin
-
Tumour-specific enzymes are hijacked to deliver anticancer drugs

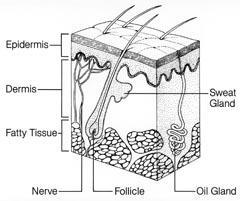
Skin cancer manifests itself in three forms: basal cell and squamous cell carcinomas or malignant melanoma. The carcinomas begin in the skin or in the cells that line internal organs. Basal cells are found in the lower part or base of the epidermis, the outer layer of the skin; squamous cells are found in the tissue that forms the surface of the skin. Malignant melanoma begins in the melanocytes, the pigment (melanin) producing cells which are evenly distributed in the basal epidermal layer of the skin (Fig 1) and which give skin its colour variations, including tanning and freckles. The prognosis for patients suffering from basal cell and squamous cell carcinomas is generally good, because these cancers rarely spread to other sites (metastasise) and form secondary tumours. Usually, the tumour is successfully removed by surgery and no chemotherapy is needed. In contrast, malignant melanoma readily spreads to secondary sites and is therefore much more difficult to treat effectively. During the early stages, diagnosis leads to treatment by surgical removal but at the moment there is no effective treatment for advanced stage melanoma so delayed diagnosis can be fatal. Although malignant melanoma accounts for just 10 per cent of all skin cancers (currently, around 132,000 melanoma skin cancers occur globally each year), it is responsible for more than 90 per cent of skin cancer deaths.1
Causes and symptoms
The two main factors that affect a person's risk of getting melanoma are whether they have a genetic disposition to the illness and excessive exposure to sunlight. Individuals from families with a history of melanoma are at significant risk of developing the disease,2 as are people who freckle or sunburn easily without tanning.3-5
Pale-skinned individuals produce less melanin than people of Asian or African origin, and are most at risk from the carcinogenic effect of ultraviolet light from the sun. Ultraviolet light is subdivided into UVA, UVB and UVC.
All UVC (very short wavelength uv light) is absorbed by the atmosphere and does not reach the Earth's surface. UVB is biologically damaging, but most of this is absorbed by the atmosphere. Long wavelength UVA is the most intense uv light reaching the Earth and penetrates furthest into tissue, but it is not as biologically damaging as UVB. The ozone layer acts as a protective filter which reduces the amount of uv light reaching the Earth's surface. Australia has the highest incidence of melanoma in the world, which is attributed to a hole in the ozone layer over the country.
The primary symptom of melanoma is usually a change in appearance of a mole, or some other form of growth, on the skin. Bleeding or any change in appearance of a coloured skin lesion is a warning sign.
Current treatments
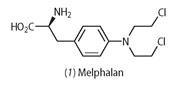
When only the epidermis is affected (melanoma in situ), the melanoma can be removed surgically with most patients enjoying a long life.6,7 If, however, the melanoma has spread, chemotherapy, using anticancer drugs such as N -mustard compounds, is necessary.8 The N-mustard drugs, such as melphalan (1), are derived from mustard gas, a chemical warfare agent used in World War I. Consequently, they show similar toxic effects to mustard gas, such as the destruction of bone marrow and white blood cells. The side effects are a result of the mode of action of these drugs: they attack cells that are undergoing division, and so normal fast-dividing cells are also affected. The N-mustards cannot cure melanoma, owing to the drug-resistant characteristics of this disease, but they do relieve symptoms and prolong life for about 9-11 months.
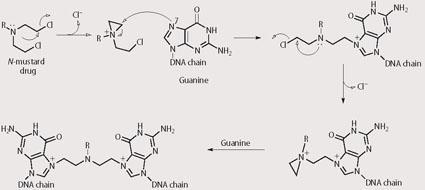
Specifically, the N-mustards work by reacting with DNA and changing its three dimensional structure by forming inter- and intra-strand crosslinks (see Scheme 1). DNA exists as a double helix structure. The helix must unwind to reveal the DNA fingerprint, which then acts as a template for DNA replication. The formation of inter- and intra-strand crosslinks prevents the helix from uncoiling and cell division of the cancer cell ceases. Melphalan9 is a phenylalanine derivative of N-mustard that exists as a clear fluid and is given intravenously to a patient through a cannula, or is administered as a tablet to be taken orally on an empty stomach.
Immunotherapy is an alternative treatment which uses the body's own immune system to fight the melanoma and has fewer side effects than chemotherapy. The approach involves administering cytokines, ie proteins that stimulate the immune system, such as interferon alpha and interleukin 2. These have been shown to shrink metastatic melanomas in some patients but have side effects such as fever, aches and severe fatigue.
Radiation therapy, which uses high-energy rays, such as x-rays, is also used to kill cancer cells. This treatment is usually used to control metastases which have spread to the brain and bones, rather than to treat primary tumours, and does relieve some of the symptoms caused by melanoma.
Prodrug therapy
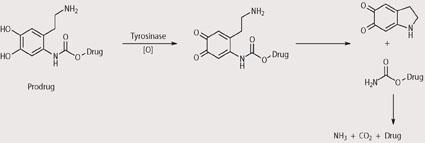
One of the major problems with chemotherapy is that the drugs used have poor selectivities for cancer cells and cause damage to normal cells and organs. For this reason, we are investigating non-toxic 'prodrugs' that are converted into the toxic drugs only at the tumour site. This could be done, for example by using enzymes that are present only at the tumour site.10 Melanomas contain high levels of the enzyme tyrosinase, which is virtually absent from other cells in the body.11
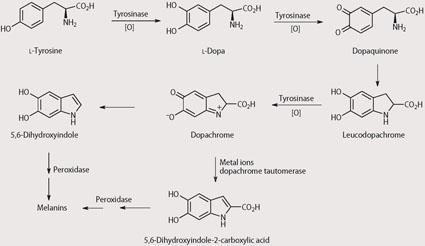
Tyrosinase is used by skin cells to synthesise melanin, see Scheme 2. Within our group we are trying to 'hijack' this tyrosinase-mediated biological pathway to convert a prodrug to an intermediate that is unstable and thus leads to release of the toxic drug. Since the tyrosinase enzyme is only located at the tumour we believe that this should allow delivery of the toxic drug selectively at the tumour, leading to minimal toxicity at other sites within the body. This approach has become known as MDEPT - melanocyte-directed enzyme prodrug therapy.
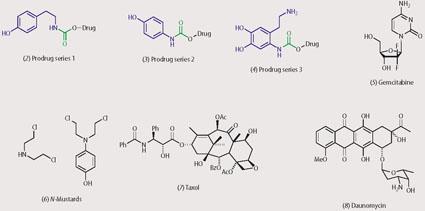
The first stage of our MDEPT programme involved the multistep syntheses of a series of prodrugs.12 We prepared three series of prodrugs (see structures (2)-(4)), each with its own tyrosinase-promoted mode of drug release. In each series the prodrugs contained a unit (derived from a phenol or catechol) that was a substrate for tyrosinase (shown in blue) and this was joined via a linking unit, shown in green, (carbamate, urea, thiocarbamate or thiourea) to a cytotoxic drug (shown in black).
The linking unit alters the chemical and electronic properties of the cytotoxic drug, so that its toxicity is temporarily masked. The tyrosinase substrate provides a handle within the prodrug for tyrosinase to act upon, to initiate drug release. We incorporated a range of cytotoxic agents within the prodrugs, eg gemcitabine (5), N-mustards (6), taxol (7) and daunomycin (8).
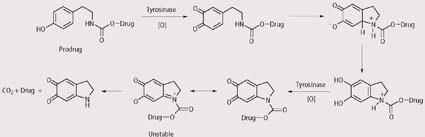
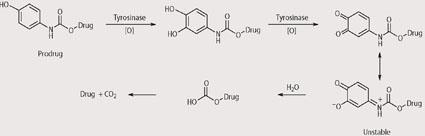
We analysed the prodrugs using a range of analytical techniques to check that they are stable in the absence of tyrosinase but release the cytotoxic drug upon exposure to tyrosinase. For example, using HPLC we have shown that the prodrugs have useful stabilities under physiological and hydrolytic conditions but release the cytotoxic drug rapidly on exposure to tyrosinase. In particular, prodrugs that contain N-mustards as the cytotoxic agents have shown promising results and this is particularly pleasing given that N-mustards are already used in the clinic to treat melanoma. Although the exact mechanisms by which the prodrugs release the cytotoxic drugs upon exposure are not known, some suggestions are given in Schemes 3-5. In each case reaction with tyrosinase is believed to produce an unstable intermediate which is hydrolysed within the cancer cell and releases the cytotoxic drug.
Although our initial results illustrated the general feasibility of this approach, we decided to try a more rigorous method for testing the value of the MDEPT approach. This involved measuring the cytotoxicity of the prodrugs in a range of cells that contain varying levels of tyrosinase. Ideally, a good correlation between the level of tyrosinase in the cell and cell death should be seen. This proved to be the case for a range of prodrugs, with those derived from series one (Scheme 3) being particularly effective. For example, relatively low toxicity was seen in tyrosinase deficient CHO and Caki-2 cell lines (IC50 = 18.9 and 45.2μM respectively), but much greater efficacy was seen in an StMI11a cell line, which shows a high level of tyrosinase activity (IC50 = 2.8μM).

Our results suggest that the prodrugs appear to function as melanocyte-directed prodrugs, liberating the toxic moiety selectively in the desired manner. Before these compounds can be tested in the clinic we hope to reduce the toxicity of the prodrugs in non-cancerous tissues to optimise the therapeutic window for these compounds. In the meantime, it is essential that primary prevention of malignant melanoma is through continuous education of the general public about the hazards of sun exposure.
Professor Helen M. I. Osborn is director of pharmaceutical chemistry in the school of pharmacy at the University of Reading, Whiteknights, Reading RG6 6AD; Samaila Jawaid is a postdoctoral researcher in the same department; and Nana Aba O. Williams completed her PhD under the supervision of Professor Osborn at the University of Reading.
Related Links
The Merck Manual of Health and Aging
Information on skin cancer deaths from the Merck Manual of Health and Aging
References
- Skin cancer deaths log on to website
- A. M. Goldstein and M. A. Tucker, Arch. Dermatol., 1998, 137, 1493.
- R. G. Langley and A. J. Sober, Cancer Invest., 1997,15, 561.
- B. K. Armstrong and A. Kricker, Dermatol. Clin., 1995,13, 583.
- R. Marks, Clin. Exp. Dermatol., 2000, 25, 459.
- A. Mota and A. Deisseroth, Cutaneous Mel., 2000, 27, 3463.
- W. Chen, S. C. Metha and D. R. Lu, Adv. Drug Delivery Rev., 1997, 26, 231.
- A. Awada and M. Piccart, Curr. Opin. Oncol., 2000, 12, 289.
- W. B. Pratt and R. W. Rudon, Anticancer drugs, Chap 5. Oxford: OUP, 1979.
- M. Rooseboom, J. N. M. Commandeur and N. P. E. Vermeulen, Pharmacol. Rev., 2004, 56, 53.
- P. A. Riley, Pigment Cell Res., 2003, 16, 548.
- S. Knaggs et al, Org. Biom. Chem., 2005, 3, 4002.



No comments yet