


Around 150 years ago potassium bromide marked the first effective treatment for epilepsy. Since then new anti-epileptics with various degrees of success have come on the market, partly through chance and partly as clinicians and researchers develop their understanding of how these drugs work.
-
The treatment for epilepsy has moved from the obscure into the realms of rational drug design

The brain is made up of billions of nerve cells, each linked by myriad connections (synapses). Chemical messenger molecules (neurotransmitters) pass between these cells, stimulating electrical impulses along their projections (axons). Regular waves of electrical activity are produced in our brains at rest, which increases on exercise or during mental activity.
However if the brain is damaged, either physically or genetically, these regular waves can become disordered, resulting in unpredictable, excessive electrical discharges from the nerve cells. At this time the person loses consciousness, often with jerking movements of limbs - this is an epileptic seizure. The condition affects ca one person in 100, a quarter of whom have their first seizure before their fifth birthday.
Mistletoe and cherry water...
In the centuries gone by, fear of the condition led to all sorts of desperate, if futile, treatments.1 Some were surgical - eg castration or removal of a circular section of bone from the skull - while others were chemical. Concoctions of mistletoe and cherry water enjoyed some popularity in the 18th century, as did, a little later, medicines based on zinc oxide or silver nitrate. Too much silver nitrate, however, caused an irreversible grey coloration of the skin, an unfortunate advertisement for the disease that it was supposed to cure.
Since seizures are intermittent, chance periods of freedom from fits could be misguidedly seen as 'proof' of a given (ineffective) treatment being a 'cure'. In fact, no treatment was effective - until almost exactly 150 years ago, when the surgeon Sir Charles Locock discovered the simple inorganic chemical, potassium bromide, to be an effective anti-convulsant. Unlike almost all drug development at the time, generally based on a chance dosing, imperfectly analysed, this advance came as a result of scientific reasoning - if founded on a misconception.
The KBr story
In 1857 the physician Edward Sieveking, at a meeting of the Royal Medical and Chirurgical Society, reported on various treatments he was working on for epilepsy. In the discussion that followed, Sir Charles Locock announced his two 'cures' for the disease. One was the extraction of over-crowded teeth (which was not followed up) and the other was the use of potassium bromide to treat women with 'hysterical epilepsy', the incidence of which was linked to their menstrual cycle.
Locock had come across a report from 'a German', who found that a sufficient dose of KBr induced temporary impotence. Locock reasoned that the chemical dampened sexual excitement, which he thought caused seizures, and went on to show that doses of KBr, ranging from 0.3-0.6g taken three times a day, rendered 14 out of his 15 patients free from seizures.2
By the late 19th century several studies of the efficacy of bromide therapy for treating other types of epilepsy had been published.3 Three of these give the following statistics: of the 873 patients treated, 200 had their seizures 'arrested' (completely controlled), 374 showed some improvement and the remainder, 299, received no benefit.
However, KBr treatment came with dose-related side effects,4 which ranged from continual sleepiness to diminished mental function, drooling, severe acne and a staggering gait. Nonetheless, these side effects were thought preferable to a lifetime of unpredictable fits. Potassium bromide remained the only effective treatment for epilepsy for the next 50 years. Any successor would have to show greater effectiveness and fewer side effects. The next part of our story results from another chance observation.
Phenobarbital
In 1864 the organic chemist Adolf Baeyer, working in Berlin, synthesised barbituric acid (1). A few years later in 1868 Adolph Strecker deduced its structure, and in 1903 Emil Fischer and Joseph von Mering discovered a simple synthesis for it, and its relatives, by condensing malonic acid (propanedioic acid), or substituted malonic acids, with urea.
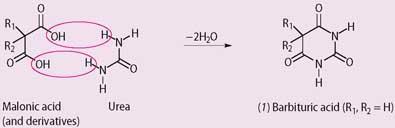
A year later chemists at Bayer discovered that when R1 = R2 = ethyl, the product (diethylbarbituric acid) induced sleepiness, and went on to market the drug as the hypnotic, Veronal. In 1912 they produced another analogue (phenobarbital, Luminal in which R1 = ethyl and R2 = phenyl), which combined Veronal's hypnotic effect with a useful sedative action. This latter aspect ought to have led to its immediate assessment as an anti-convulsant drug, but this was not the case. Phenobarbital's action against epilepsy was discovered accidentally.
Alfred Hauptmann, a young psychiatrist working in Hamburg-Eppendorf, decided to use phenobarbital to sedate some of his epileptic patients:2
"I should draw attention to the susceptibility of epileptic attacks to Luminal. This became apparent when using this new medication as a tranquilliser, also with epileptic patients.... To date there has been no mention in published articles about Luminal and its application in the treatment of epilepsy.... The action upon the attacks was two-fold: the frequency of the attacks abated appreciably, or the attacks stopped altogether. In the less severe cases the nature of the attacks changed. Severe attacks (involving) tongue-biting etc were replaced by attacks of shorter duration or brief faints."
Phenobarbital went on to become the drug of choice for the treatment of epilepsy until the early 1940s. However, the search for alternative anti-convulsants continued, spurred on by three weaknesses of phenobarbital:
-
as Hauptmann indicated, some patients (more recent research suggests ca 20 per cent) still suffered from seizures, despite the treatment;
-
at higher (but sometimes necessary) doses, sleepiness became a problem. This could be reduced by taking the main fraction at bedtime, but it was sufficient to stop the patient driving or working with machinery;
-
phenobarbital's therapeutic index (TI)* is low (ca 10). This means that lethal overdose, either accidental or deliberate, was a significant risk.
(*This is the lethal dose of a drug for 50 per cent of the population divided by the minimum effective dose for 50 per cent of the population. Drugs with high TIs are preferred to those with low TIs.)
Animal studies
In the mid-1930s US researchers Houston Merritt and Tracy Putnam used an animal model (cats) to screen new compounds that might control and prevent seizures. They knew that an electric current applied across the brain would induce a fit, so they passed a current between the scalp and mouth of these unfortunate animals.5 They established the minimum current necessary to induce a fit and found this remained more or less constant for an individual test animal (the convulsive threshold).
The animal would then be given the test drug and the convulsive threshold re-determined. If this were significantly raised (suggesting the drug was preventing seizures at previously convulsive levels of brain electrical stimulation), then they would investigate the drug further. Because they knew that both potassium bromide and phenobarbital were sedatives, they tested similar chemicals. Putnam describes his approach:
"I combed the Eastman Chemical Company's catalogue, and other price lists, for suitable phenyl compounds that were not obviously poisonous. I also wrote to major pharmaceutical firms, asking if they had available, or could make, suitable chemicals. The only one of them that showed any interest was Parke, Davis and Company. They wrote back to say they had on hand 19 different compounds analogous to phenobarbital, and that I was welcome to them."
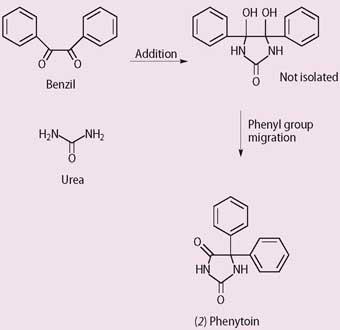
Among the 19 chemicals was a hydandoin, first synthesised by Heinrich Biltz in 1908 and again in 1923 by Arthur Dox in his company's search for patentable hypnotics (Scheme 2). However, the hydandoin didn't have the desired effect, and was put into store. Dr Oliver Kamm, Parke Davis's director of research, takes up the story and quotes from his memorandum to Dox, dated 3 April 1936:
"Putnam expressed an interest in cooperating with us in a search for a barbituric acid hypnotic of the Luminal type. He expressed the opinion that certain substances, though rejected as hypnotics, might possess anticonvulsant activity."2
The first substance listed by Parke Davis, and tested by Putnam and Merritt, was 5,5-diphenylhydantoin (phenytoin, 2). This had structural similarities to phenobarbital. Animal studies showed this substance was an anti-convulsant but, unlike pheno-barbital, was not sedating. Successful human trials were reported at the 1938 meeting of the American Medical Association.
The diphenylhydantoin (later christened Dilantin Sodium, phenytoin or Epanutin) was effective against a wider range of epilepsies (though not spectacularly so) and, with a higher therapeutic index, was ca 2.5 times less toxic than phenobarbital. But the drug also produced side effects, including nausea and vomiting, dizziness, skin rashes and a coarsening of the features of the face, with associated excessive hair growth and acne. The search for the ideal anti-epileptic drug continued.
Ethosuximide
While the drugs available up to this point were effective for many forms of seizures, they were less useful for a particular type of epilepsy, ie absence seizures, which affects children. In a typical absence seizure the child becomes unaware of their surroundings for relatively brief but frequent periods, which are disruptive and in some circumstances, such as walking along a road, dangerous.
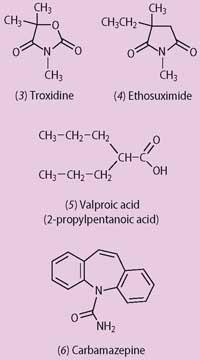
In the late 1930s chemists at Abbott Laboratories in Chicago were looking for an alternative to the common analgesic, aspirin, based on oxazolidine-2,4-diones. While testing one of their compounds, troxidone (3), the researchers discovered that it inhibited the convulsions in mice induced by high doses of the drug amolanone, a local anaesthetic agent. Troxidone was to prove to be particularly effective against absence seizures. And though it too produced side effects, including night-blindness and double vision, it showed that it was possible to treat this form of epilepsy and provoked a search for better alternatives.
Parke Davis took up the challenge and investigated over 1000 heterocyclic amides. Most promising was a near-relative of troxidone, surprisingly overlooked by Abbott. Parke Davis's drug - ethosuximide (3-ethyl-3-methyl-2,5-pyrrolidine) (4) - first synthesised in 1927, entered clinical practice in 1958. Its side effects are better tolerated than those of troxidone and it remains the treatment of choice for uncomplicated absence seizures.
Sodium valproate
During the first half of the 20th century there was considerable interest in organic compounds of bismuth as drugs. This may have been stimulated by Paul Ehrlich's introduction of the arsenical derivative Salvarsan in 1909, which revolutionised the treatment of syphilis.
However, many of the bismuth compounds were stubbornly insoluble, making it difficult to turn them into effective medications. For example, bismuth salicylate, an injection treatment for syphilis that was still in occasional use as a post-war adjunct to penicillin therapy, had to be administered as a suspension in oil.6 In a search that would render some of the bismuth compounds more soluble, the French pharmaceutical company Berthier turned its attention to valproic acid (2-propylpentanoic acid) (5) which was, apart from a 1940s investigation as to its possible esterification with glycerol to form war-time substitute fats, little studied.7
Berthier chemist Hélène Meunier found that valproic acid had a useful solubilising effect on the bismuth compounds she was exploring, but nothing initially came of the observation, presumably because of its corrosive/reactive nature. In 1961 Pierre Eymard, working in the Berthier laboratories, was trying to make derivatives of khelline, a furanochromone isolated from the Mediterranean plant Ammi visnaga, extracts of which appeared to relieve the pain of angina. But many of his compounds were barely soluble.
At Meunier's suggestion, Eymard tried using valproic acid as a solvent and found it to be effective. Consequently, he sent some of his derivatives for pharmacological testing and, remarkably, they all showed similar anti-convulsant activity. This was soon tracked down, not to his khelline products, but to the valproic acid solvent. Rather than using the corrosive acid, he used the sodium salt for clinical trials, which were published in 1963.
Sodium valproate turned out to be fairly well tolerated, though it can cause acute liver failure, particularly in children, and can affect the bone marrow, particularly the production of platelets, which are essential for clotting. Other troublesome, if not fatal, side effects include weight gain and partial, usually transient, hair loss. This can lead some patients, particularly adolescents, to abandon their treatment, sometimes with disastrous results. Nonetheless, sodium valproate is widely considered the drug of choice for the first-line treatment of generalised seizures.
Other anti-epileptics
Despite the increasing range of anti-convulsants described so far, including the advent of the benzodiazepines8 from 1960 in the management of acute seizures, there has remained no ideal anti-convulsant that can control all seizures without significant side effects.
During the 1970s carbamazepine (5H-dibenzazepine-5-carboxamide) (6) (originally marketed for the treatment of a particular painful condition of the face, trigeminal neuralgia) was recognised as an important anti-convulsant, and the 1990s saw the first use of a wide range of new agents. Even so, not everyone responds to a given medication, and some people continue to suffer seizures despite the use of two or more drugs. While most receive full or substantial benefit, about a quarter of people with epilepsy gain only limited benefit from their anti-convulsant treatment.9
Future directions
Increasing understanding of how these drugs work, eg via their effects on sodium and calcium channels, has helped more scientific development of new medicines. However, the real challenge is to prevent development of epilepsy, rather than to control the condition once it exists. The rare genetic forms of epilepsy, in which an error in genetic code for the sodium or other ion channels leads to epilepsy, offers one insight into how epilepsy may develop and the potential for drug, or other molecular, interventions.
Sometimes epilepsy can develop some years after an insult to the brain, such as a head injury. Greater understanding of how this initial injury develops at a cellular and molecular level may also offer clues as to how to intervene to arrest this process, so the original injury never progresses to cause this very troublesome condition.
The explosion of understanding in neuroscience since the early 1990s offers real hope of continuing major progress in the treatment, and even the prevention, of this age-old condition - without resorting to some of the random heroics of the past, or Sir Charles Locock's suggestion of a dental cure!
Alan Dronsfield is emeritus professor of the history of science in the school of education, health and sciences at the University of Derby, Derby DE22 1GB. Pete Ellis is professor of psychological medicine at the School of Medicine and Health Sciences, University of Otago, Wellington, PO Box 7343, Wellington South, New Zealand.
Further Reading
The most comprehensive account of the discovery of effective anti-epilepsy drugs is given in the text by Scott, and most of the quotations in this article are taken from this source.2 The earlier, and mainly bizarre, remedies are described in the book by Temkin.1
References
1. O. Temkin, The falling sickness - a history of epilepsy, 2nd edn. Baltimore: John Hopkins, 1971.
2. D. F. Scott, The history of epileptic therapy. Carnforth (UK): Parthenon, 1993.
3. W. E. Turner, Epilepsy - a study of the idiopathic disease. London: Macmillan, 1907.
4. A. T. Dronsfield, T. M. Brown and P. M. Ellis, Educ. Chem., 1998, 35 (6), 168.
5. Pioneers in epilepsy research: see link to website.
6. Diagnosis and treatment of the venereal diseases: see link to website.
7. J. W. A. Meijer et al in Discoveries in psycho- and neuro-pharmacology, M. J. Parnham and J. Bruinvels (eds). Amsterdam: Elsevier Science, 1983.
8. A. T. Dronsfield and P. M. Ellis, Educ. Chem., 2008, 43 (6), 148.
9. BMJ best treatments for epilepsy: see link to website.





No comments yet