


Entropy is probably the most misunderstood of thermodynamic properties. While temperature and pressure are easily measured and the volume of a system is obvious, entropy cannot be observed directly and there are no entropy meters. What's the solution?
-
Does it really make sense to describe entropy in terms of the disorder of a system?
-
Entropy is dynamic - something which static scenes don't reflect
Thermodynamics deals with the relation between that small part of the universe in which we are interested - the system - and the rest of the universe - the surroundings. Thermodynamic properties depend on the current state of the system but not on its previous history and are either extensive - their values depend on the amount of substance comprising the system, eg volume - or intensive - their values are independent of the amount of substance making up the system, eg temperature and pressure. Entropy is a thermodynamic property, like temperature, pressure and volume but, unlike them, it can not easily be visualised.
Introducing entropy
The concept of entropy emerged from the mid-19th century discussion of the efficiency of heat engines. Generations of students struggled with Carnot's cycle and various types of expansion of ideal and real gases, and never really understood why they were doing so.
Many earlier textbooks took the approach of defining a change in entropy, ΔS, via the equation:
ΔS = Qreversible/T (i)
where Q is the quantity of heat and T the thermodynamic temperature. However, it is more common today to find entropy explained in terms of the degree of disorder in the system and to define the entropy change, ΔS, as:
ΔS = -ΔH/T (ii)
where ΔH is the enthalpy change. This more modern approach has two disadvantages. First the units of entropy are Joules per Kelvin but the degree of disorder has no units. Secondly, the equation (ii) defining entropy change does not recognise that the system has to be at equilibrium for it to be valid.
We prefer to consider that the entropy of a system corresponds to the molecular distribution of its molecular energy among the available energy levels and that systems tends to adopt the broadest possible distribution. Alongside this it is important to bear in mind the three laws of thermodynamics. The first law deals with the conservation of energy, the second law is concerned with the direction in which spontaneous reactions go, and the third law establishes that at absolute zero, all pure substances have the same entropy, which is taken, by convention, to be zero.
In classical terms, systems at absolute zero have no energy and the atoms or molecules would be close packed together. As energy, in any form is supplied to a system, its molecules begin to rotate, vibrate and translate, which is observable as a rise in temperature. The rise in temperature caused by a given quantity of heat will be different for different substances and depends on the heat capacity of the substance.
Since different substances have different heat capacities and because some compounds will have melted or vaporised, by the time they have reached their standard states at 298 K, their standard entropies will be different. For example, the standard entropy of graphite is 6 J K-1 mol-1, whereas that for water it is 70 J K-1 mol-1, and for nitrogen it is 192 J K-1 mol-1.
The distribution of energy
To obtain some idea of what entropy is, it is helpful to imagine what happens when a small quantity of energy is supplied to a very small system. Suppose there are 20 units of energy to be supplied to a system of just 10 identical particles. The average energy is easily calculated to be two units per particle. Although this is the average energy, this state of the system can only arise if every particle has two units of energy. If all the energy were concentrated in one particle, any one of the 10 particles could take it up and this state of the system is 10 times more probable than every particle having two units each.
If the energy is shared between two molecules, half of the energy could be given to any one of the 10 particles and the other half to any one of the remaining nine particles, giving 90 ways in which this state of the system could be achieved. However, since the particles are indistinguishable, there would be only 45 ways in which this state can be achieved. Sharing the energy equally between four particles increases the number of possible ways this energy can be distributed to 210. This kind of calculation quickly becomes very tedious, even for a very small system of only 10 particles, and completely impracticable for systems where the number of particles approaches the number of atoms/molecules in even a very small fraction of one mole.
The very large numbers involved allow a number of simplifying approximations to be made and Ludwig Boltzmann was able to show that as the energy is spread more widely in a system, the number of possible distributions increases to a peak - corresponding to the most probable distribution - and then declines (see Fig 1).
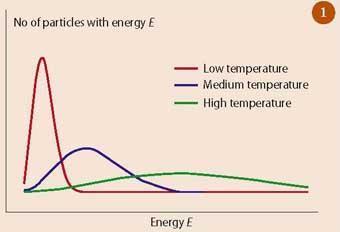
At low temperatures, when the system has only a small amount of energy, comparatively few energy levels can be explored by the molecules because there is simply not enough energy in the system. Most of the molecules will have close to the average energy but there will be some with more than the average energy which occurs because there are some other molecules which have less than the average amount of energy.
As energy is supplied to the system, higher energy levels can be explored and the peak shifts towards higher energies. The temperature rises but, because the amount of substance in the system is constant, the height of the peak decreases as the particles with the most probable energy take up more energy and move into these higher energy levels. This spreading out of energy into a variety of energy levels is an essential characteristic of entropy.
Another important feature of entropy is that it is dynamic - although the amount of energy and amount of substance in an isolated system is constant - the energy is constantly being exchanged between different energy levels as a result of the collisions between molecules. This is in direct contrast to the common analogy used to describe entropy - ie the chaos of a teenager's room is often quoted as a system with high entropy, but the socks under the bed never exchange energy with the shirt hanging on the bed-post.
Standard entropy
When considering calculations, it is important to remember that the defining equation (i) requires the quantity of heat to be taken up reversibly. (This is often overlooked by most textbooks written for A-level specifications.) When a system, initially at 0 K, absorbs this quantity of energy, its temperature rises by δT to 0 + δT or T1. A further quantity of energy, q, brings about an entropy change, as defined by equation (i), and the temperature rises to T1 + δT or T2. This process of adding energy to the system by a series of reversible absorptions eventually brings the system to 298 K and the sum of these operations gives the standard entropy of the system (which is the difference between the entropy of the substance in its standard state and its entropy at 0 K).
Rather than tediously adding these minute increments, the use of calculus allows us to achieve the same result. Entropy changes accompanying phase changes - eg melting, vaporisation etc - can be calculated without the use of calculus if the two phases are in equilibrium (ie at the melting point or boiling point). Under these conditions, the entropy change can be calculated from equation (ii).
Tables of standard entropies list the entropy of 1 mole of an element or compound in its standard state at 298 K - the standard state of a solid element or compound is the pure solid, that of a liquid is the pure liquid in equilibrium with its vapour and that of a gas is when it is at a pressure of 105 N m-2 or 1 standard atmosphere.
The use of an increase in total entropy as a criterion of spontaneity of reaction has to be treated with some caution. Its chief disadvantage is that it requires knowledge of the entropy changes in both the system and its surroundings.
ΔStotal = ΔSsurrounding + ΔSsystem
Moreover, since the standard entropies relate to standard states at 298 K, it is not appropriate, in most cases, to use these values at different temperatures. Whereas data can generally be obtained directly from the system itself, this is not the case with the surroundings which are generally assumed to be large enough not to be significantly affected by the changes occurring in the system.
Thermodynamics is an elegant and powerful tool but it has to be used with caution. Chemical reactions involve reactants and products in reactions which may not necessarily go to completion. Why this should be so involves both the enthalpy and entropy changes as reactants turn into products.
Exothermic reactions result in less energy in the system but are usually (not always) associated with a reduction in the entropy of the system, endothermic reactions are generally the reverse of this. When there are two opposing changes, a balance point has to be attained and the result is a mixture of reactants and products.
The key to determining which way and how far a chemical reaction will go is provided by another thermodynamic property - the Gibbs free energy (G) and it should come as no surprise to find that it is related to both the enthalpy and the entropy of the system:
ΔG = ΔH - TΔS
When the concept of entropy is being introduced, it is important that misconceptions should be avoided, particularly the idea that entropy represents the degree of disorder in the system. Entropy is dynamic - the energy of the system is constantly being redistributed among the possible distributions as a result of molecular collisions - and this is implicit in the dimensions of entropy being energy and reciprocal temperature, with units of J K-1, whereas the degree of disorder is a dimensionless number.
Lawson Cockcroft is retired and can be contacted at 35 Clairevale Road, Heston, Middlesex TW5 9AF; Graham Wheeler teaches chemistry at Reading School, Erleigh Road, Reading RG1 5LW.





No comments yet