


Analgesics, ie pain-relieving drugs, fall into two categories: those that also reduce body temperature in fevers (antipyretics), and those that act mainly on the brain - typically morphine and diamorphine/heroin. Here we consider members of the first group, particularly those once designated 'coal tar analgesics'. Paracetamol, our most popular over-the-counter pain killer, is one of these.

Towards the middle of the 19th century, a person suffering from mild to moderate pain, eg headache, toothache, or arthritis, had little choice of remedies. Narcotics, typically laudanum, an ethanolic extract of crude opium, were available but were unpopular because they had side effects - ie sleepiness, constipation and the risk of addiction. Salicylic acid, at this time only available as derivatives from willow bark, became freely and cheaply available after the synthesis of the free acid by Hermann Kolbe in 1859, but was toxic. Its further development into the well-tolerated and still widely used acetyl derivative, aspirin, did not occur until the late 1890s.1 So, in the second half of the 19th century there was a need for more acceptable analgesics for everyday use.
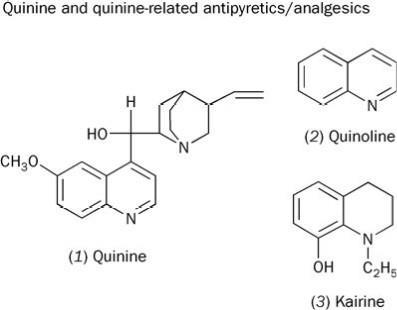
From quinine to quinoline
Quinine had been used in the West since 1640 to treat the high temperature, fever and headache caused by malaria. In 1842, Charles Gerhardt showed that quinine (1), on fusion with KOH, yielded quinoline (2). Within a few years quinoline was becoming cheaply available from coal tar distillation. It was a short step to see if quinoline was an analgesic/antipyretic.
Despite its nauseating taste and smell, quinoline was occasionally used until ca 1900 with some success against typhoid fever, acute rheumatism and, as its tartrate salt, against neuralgia (a severe nerve pain, often of the face or head). However, in a few individuals quinoline caused cyanosis, a bluish coloration of the skin brought on by an inadequate supply of oxygen and often associated with heart disease. The limited success of this compound prompted chemists to find a quinoline derivative that retained the therapeutic effect of quinoline but was less toxic. Several compounds were synthesised, including the partially hydrogenated quinoline derivative, kairine (3), introduced into medical practice from 1882.2 Described as a powerful antipyretic, kairine sometimes caused profuse sweating and vomiting. Also, like quinoline, there was a risk of cyanosis for some patients. Although better than the parent compound, kairine was far from perfect.
Emil Fischer's influence
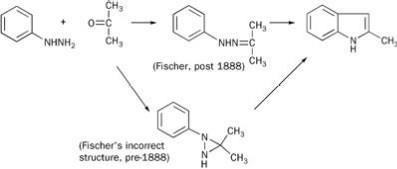
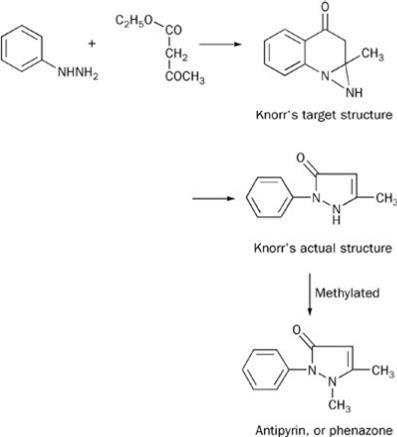
In 1883 Emil Fischer discovered, serendipitously, phenyl hydrazine, and used it in his indole synthesis of 1883 (see Scheme 1). (At the time of this work Fischer believed that the intermediate hydrazone was based on a N-N-C ring, though this later proved incorrect.) Fischer's assistant, Ludwig Knorr, sought to extend this synthesis to make a quinoline derivative. The chemistry he proposed - a pre-1888 Fischer-type intermediate coupled with an indole-like reconnection to the 2-position of the parent benzene ring - is shown in Scheme 2. His target product had some structural features of kairine-type antipyretics, so he sent it for medical evaluation. More or less simultaneously he showed that his proposed structure was wrong and that he had made a phenyl-substituted pyrazolone,2 but the testing went ahead.
The product had neither antipyretic nor analgesic action. Professor Wilhelm Filehne, who evaluated the compound for its pharmacological properties, suggested alkylating the NH group, thus making the structure more closely resemble that of kairine. This Knorr did, and his methylated product, Antipyrin or phenazone, was soon shown to be both an antipyretic and an analgesic. Its success is described by medical author Walter Sneader:
Five years later (after its synthesis) it was put to the test when a major influenza epidemic swept through Europe. For the next 15-20 years Antipyrin was the most widely used drug in the world, until aspirin began to outsell it. At the turn of the century Hoechst Dyeworks was producing upwards of 17000 kilos annually. If any single product can be said to have established the commercial viability of synthetic drugs, it was surely phenazone.3
However, the use of Antipyrin was not without risk; in some people it caused agranulocytosis, ie suppression of bone marrow with the consequential loss of white cells. This left the patient vulnerable to infection, which was often fatal.
Scheme 1 Fischer's route to 2-methylindole
Scheme 2 Knorr's route to Antipyrin
A dispensing error
The next chapter of the story is founded on a dispensing error.3 Professor Adolf Kussmaul of the University of Strassburg was testing a variety of products against intestinal worms. One of these was naphthalene, another coal tar product. His two young assistants, Arnold Cahn and Paul Hepp, had little success with this until they treated a patient suffering from a multitude of complaints alongside worms. They noted that the 'naphthalene' had a surprising yet unreported antipyretic effect. Further investigation, however, revealed that the pharmacy had supplied their patient, not with naphthalene, but with acetanilide, a solid with a near-identical crystalline appearance. Cahn and Hepp published their discovery in 1886, cryptically attributing it to 'a fortunate accident',4 and the following year acetanilide was manufactured as Antifebrin. The latest antipyretic was effective, remarkably cheap, and presumably, to the annoyance of the manufacturers of Antipyrin, well named. Although its use continued in a few over-the-counter preparations until 1971, Antifebrin was soon replaced by other, safer analogues, notably phenacetin, first synthesised in 1887.
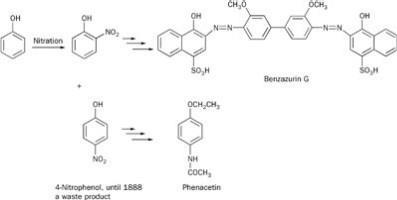
In the late 1880s the dyestuffs manufacturer Friedrich Bayer & Co was looking to diversify its commercial interests and to find a use for 4-nitrophenol, a side product available in vast amounts from its synthesis of the blue dye Benzazurin G (Scheme 3). Research director, Carl Duisberg, challenged his team to come up with an exploitable use for this compound. One of them, Oscar Hinsberg, found that, in three steps - ie reduction of NO2 to NH2; ethylation of the OH group; and finally acylation of the NH2 group - he had made a well-tolerated antipyretic and analgesic, which possessed all the advantages of Antipyrin and Antifebrin and (apparently) none of their disadvantages. His new product, phenacetin, enjoyed much popularity for almost a century as an over-the-counter remedy (often combined in tablets with caffeine and aspirin) for headaches and hangovers. However, in the 1960s evidence was mounting to suggest that phenacetin might be causing renal failure and renal tumours in some heavy users. In 1980 phenacetin was banned in the UK.
Scheme 3 Bayer's route to the blue dye Benzazurin G
Paracetamol, lost and found
Until relatively recently drug discovery was very much a 'hit and miss' process. Enormous numbers of organic chemicals were synthesised and tested against a whole raft of diseases. When one showed some promise, then its homologues and near-relatives would be explored to see if they were more effective. It was in this spirit, and to find a better alternative to phenacetin, that N -(4-hydroxyphenyl)ethanamide (4) (later named paracetamol) was discovered and evaluated in 1893.
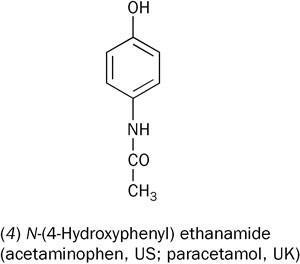
The clinical trials of this drug were carried out by Germany's leading physiologist, Joseph von Mering in collaboration with chemists at the Bayer Company. Von Mering was unhappy with the results of his clinical trials because, while it was an effective analgesic and antipyretic, in some cases its use was associated with the development of methaemoglobinaemia. This is a form of cyanosis caused by oxidation of the Fe(II) in haemoglobin to Fe(III), making iron unavailable for oxygen transport. The drug-induced form is rarely fatal (assuming medication is stopped), though the symptoms (blue lips and darkening of the skin) are alarming to the patients and their carers. Today it is thought that von Mering was observing the clinical effects of a toxic impurity (possibly 4-aminophenol) in his preparation, rather than those of the acylated target compound.3 However, his observations and his stature as Germany's leading physiologist meant that investigation of paracetamol was abandoned for over half a century.
Paracetamol rediscovered
That a compound as simple as acetanilide was effective as an analgesic and antipyretic prompted 20th century investigations into its possible mode of action. Was its efficacy, for example, the result of initial hydrolysis, yielding aniline itself?
As the century progressed, developments in colorimetric methods of quantitative analysis enabled monitoring of the body fluids of human volunteers dosed with therapeutic amounts of acetanilide. In 1947 Yale physiologists David Lester and Leon Greenberg reported their analyses of blood, plasma and urine of individuals who had received 1g doses of acetanilide. They found:
- the major metabolites in blood and plasma were N-acetyl-p-aminophenol and its 'hydroxy conjugates' (the acylated aminophenol esterified with sulphuric or gluconuric acid) in ca 1:1 proportions;
- the major metabolites in urine were the conjugates, rather than the free acylated aminophenol, in a ratio ca 25:1.
This led them to conclude:
The major metabolism of acetanilide is its oxidation first to N -acetyl-p -aminophenol with slow urinary excretion of this substance and the conversion to its hydroxy conjugate with rapid elimination.5
This work was immediately seized upon by Bernard Brodie and Julius Axelrod who were working in the New York University College of Medicine. They confirmed the ubiquity of the acylated aminophenol and continued:
The high concentration of this metabolite in the plasma prompted an appraisal of its analgesic effect. Studies on human subjects . showed N-acetyl-p-aminophenol to be, dose for dose, equal in analgesic activity to acetanilide (and) when administered orally, was not attended by the formation of methaemoglobin, nor at least in vitro, did it destroy red cells. It is possible, therefore, that it might have distinct advantages over acetanilide as an analgesic.6
Commercial production
Coincidentally, with the rediscovery of paracetamol, James Roth, a US gastroenterologist, was publicising his concerns about the adverse effects of long-term use of aspirin - ie stomach irritation to the point of bleeding and a reduced ability of the blood to clot. With the publication of Brodie's paper, Roth became an advocate of paracetamol as a safer alternative to aspirin. Roth was also principal consultant to McNeil Laboratories, a Philadelphia-based drug company, and not surprisingly perhaps the company shared his interest.7 By 1951 it had identified a need for an analgesic/sedative preparation that could be made available on a prescription-only basis and within two years it had introduced Algoson, a preparation containing paracetamol together with sodium butabarbital, a sedative.
In 1955, the success of Algoson prompted McNeil Laboratories to introduce Tylenol Elixir, a medicine for children which contained paracetamol as its sole active ingredient. Some years later, in 1961, the company marketed paracetamol tablets (also called Tylenol) as an over-the-counter preparation.8One year later, in the UK, Sterling Winthrop marketed paracetamol as Panadol, initially as a prescription product. On the expiry of the UK patent rights in 1963 paracetamol was included in the British pharmacopoeia, and became a generic medicine which could be manufactured by any pharmaceutical company. It later became even more widely available as an over-the-counter non-prescription analgesic.8
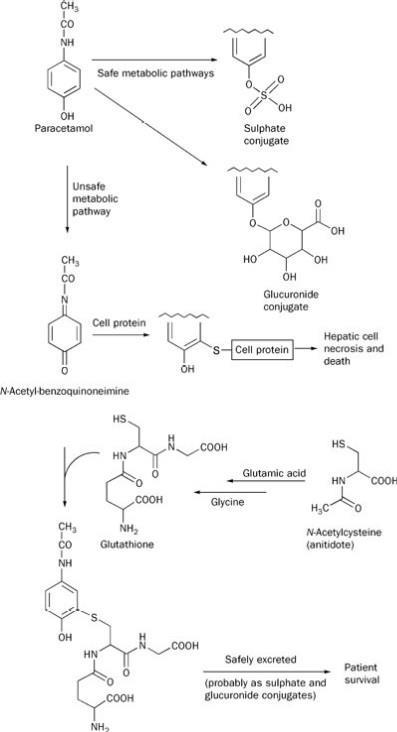
Roth looked forward to a secure place in the pharmacopoeia for paracetamol based on its lower toxicity than aspirin. However, the recurring theme in clinical pharmacology is that the wonder drug of the day often proves to have at least one Achilles' heel, if not several. Over 50 years on, the limitations of paracetamol are now recognised. In overdose, paracetamol can cause serious and sometimes fatal liver damage (Scheme 4). When phenacetin was under close scrutiny for its adverse effects on the kidney, paracetamol was found to increase the risk of renal damage too, though to a lesser extent.
Currently there is some concern about the effects of long-term regular use of high doses of paracetamol on the liver. However, it is the recognition of the limitations of existing compounds and a clearer understanding of their actions and side effects that drives the development of new agents. Just as the COX-2 inhibitors such as Celebrex and Vioxx came from an increased understanding of the therapeutic and undesirable actions of aspirin, further understanding of the actions and side effects of paracetamol may also lead to new and safer painkillers. Whether the base compounds for these agents will come from the distillation of coal tar, or from de novo synthesis, only time will tell.
Scheme 4 'Safe' and 'unsafe' metabolic pathways of paracetamol
Alan Dronsfield is professor of the history of science in the school of education, health and sciences, at the University of Derby, Kedleston Road, Derby DE22 1GB. Trevor Brown is professor of applied materials chemistry at the same institution. Peter Ellis is professor of psychological medicine at the Wellington School of Medicine and Health Sciences, University of Otago, PO Box 7343, Wellington South, New Zealand.
The mode of action of paracetamol

In the past, paracetamol has been classified as a non-steroidal anti-inflammatory drug alongside the ubiquitous aspirin and later agents with more potent anti-inflammatory action, such as indomethacin. For many years the mechanism of action of all these drugs was unknown. However, in 1971 Sir John Vane found that aspirin inhibited an enzyme, cyclo-oxygenase (COX), which was crucial to the conversion of phospholipids to prostoglandins and thromboxanes. These are responsible for an increased blood flow and other changes during inflammation, leading to swelling and pain in the local tissues.1This seemed to account for these drugs' actions as analgesics, antipyretics, anti-platelet and anti-inflammatory agents and also their side effects, ie ulceration of the stomach. Parcetamol is thought to interfere with the 'household maintenance' responsible for repairing the stomach lining, inevitably damaged by stomach acid.
The next year (1972) Vane reported that paracetamol inhibited COX activity in dog brain homogenate, but not in spleen-derived material. The implication was that paracetamol operated directly on the brain, rather than at the local site of the pain. However, he used high doses of paracetamol in his dogs. At standard human doses paracetamol seemed to have little effect on cyclo-oxygenase (COX) - leaving the action of paracetamol unexplained. More recently, however, the COX hypothesis for paracetamol's action (on the brain) has received experimental support.9
Paracetamol overdoses
In 2001 the number of deaths from paracetamol overdoses was 99.8The drug is metabolised in the liver. In therapeutic dosages most of it ends up as the sulphate or glucuronide, which are then harmlessly excreted in the urine. But in larger, potentially toxic doses, some of it is metabolised to N-acetyl-benzoquinoneimine (Scheme 4).11 This highly reactive species can be inactivated by reaction with glutathione but when stores of this are used up, the imine attacks cell proteins, destroying the liver and causing death. If the antidote, N-acetylcysteine, can be given before liver damage occurs, this can be avoided. It provides a source of cysteine to allow the body to produce more glutathione, restore its protective mechanism and rescue the person from a potentially fatal outcome of the overdose.
Acknowledgements and further reading: the pre-1950 history of analgesics and antipyretics is described in the text by Dyson and May.2 We are grateful to Dr Geoffrey Brandon of the Paracetamol Information Centre for helpful comments on this article. The Information Service publishes a most useful website on paracetamol.8 This includes information on its history and medical aspects, including the issue of 'over-dosing' with paracetamol. Readers wanting to undertake practical work in the synthesis and analysis of paracetamol are invited to consult the RSC's publications on this topic.10
References
1. T. M. Brown et al, Educ. Chem. 1998, 35 (2), 47.
2. G. M. Dyson and P. May, May's chemistry of synthetic drugs, 5th edn. London: Longmans, 1959.
3. W. Sneader, Drug discovery. New York: John Wiley & Sons, 1985.
4. A. Cahn and P. Hepp, Centralblatt Klin. Med., 1886, 33. 561.
5. D. Lester and L. A. Greenberg, J. Pharm. Exper. Therapy, 1947, 90, 68.
6. B. B. Brodie and J. Axelrod, J. Pharm. Exper. Therapy, 1948, 94, 29.
9. D. L. Simmons et al, Proc. Nat. Acad. Sci. USA, 2002, 99, 1392; J. M. Schwab et al, The Lancet, 2003, 361, 981.






No comments yet