









This article looks at developing a natural product herbicide, thaxtomin A. It will help you understand the research the journal article is based on, and how to read and understand journal articles. The research article was originally published in our Organic & Biomolecular Chemistry journal.
Natural product herbicide (±)-thaxtomin A
A one-pot multicomponent coupling/cyclization for natural product herbicide (±)-thaxtomin
Click here for the full article in the journal of Organic & Biomolecular Chemistry
Authors: Jean Paul Bourgault, Amarendar Reddy Maddirala and Peter R. Andreana
Why is this study important?
The use of herbicides in the years following World War II has proven to be more effective and less costly than hand removal or tillage for weed control. Furthermore, it is estimated that if these chemicals were discontinued the annual U.S. crop production would decrease by 20%.1 The necessary control of unwanted foliage has been a tale of two stories; on one hand there is the constant threat of herbicide resistance and on the other there are concerns regarding environmental effects.2
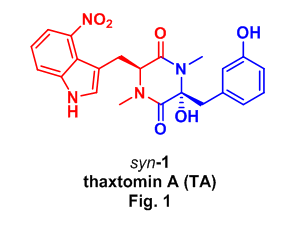
Naturally occurring thaxtomin A (TA, syn-1) was first isolated in 1989 from Streptomyces scabies3, the soil bacteria responsible for common scab disease of potatoes, and has since attracted considerable attention from industrial4 and academic5 communities as the molecule responsible for scab infections of potatoes and additionally as a potent phytotoxin demonstrating activity similar to known cellulose inhibitors. TA has also been used to control weed growth and germination in the presence of grasses and cereals causing no toxicity to these crops.4a
This interest is highly motivated by the environmentally benign and biodegradable properties of TA, which contains a cyclic dipeptide or 2,5-diketopiperazine (DKP) core, composed of a (S)-N-methyl-4-nitrotryptophan and (R)-2-hydroxy-3-(3-hydroxyphenyl)-N-methylalanine (Fig. 1). In spite of this attention there exists only a single report on the preparation of thaxtomin A,6 so we expanded on this methodology using the Ugi reaction as an alternative approach for the rapid assembly of the hydroxy DKP TA.
Further information
- A review paper on 'Phytotoxins produced by microbial plant pathogens'
- Selected further reading from RSC books:
- Enantioselective Multicatalysed Tandem Reactions
- Chemistry in the garden
What is the objective?
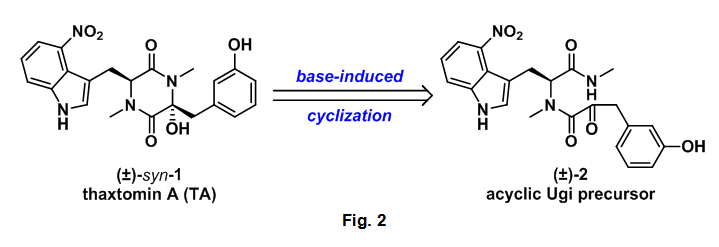
The authors' aim was to develop a one pot synthesis of TA using the Ugi reaction for the preparation of an acyclic dipeptide which could then undergo a base mediated amide-ketone cyclization to the desired diketopiperazine (DKP).
What was their overall plan?
- Utilize knowledge obtained from a Diversity-Oriented Synthesis (DOS) mantra and in the forward direction synthesize TA from the acyclic Ugi precursor, 2.
- Complete an Ugi based DOS analysis of the dipeptide (2) to the required aldehyde (3), amine (4), carboxylic acid (5), and isocyanide (6) starting materials, Fig. 3
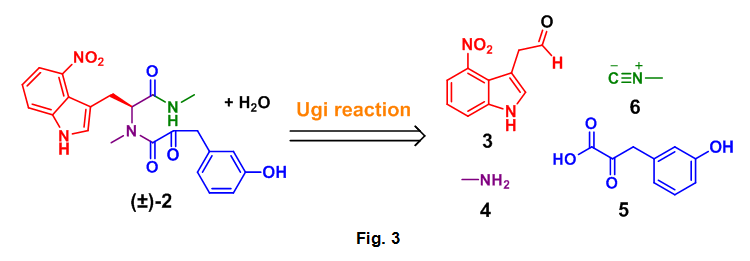
This analysis is based on the general mechanism for the Ugi reaction as seen below, Fig 4. A close examination of this reaction process allowed for the correct selection of starting materials required for the formation of 2. By following the color coded starting materials through this mechanism it can be seen how the authors arrived at 3, 4, 5, and 6.
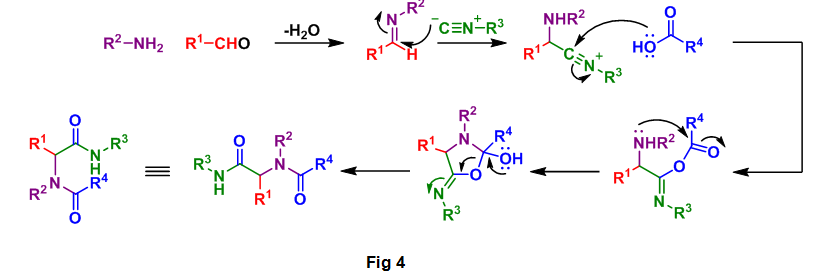
Search the literature for previous reports on the preparation of 3, 5, and 6 or derivatives thereof. Methyl amine (4) is inexpensive through commercial sources.
Prepare the starting materials based on these reports. This is not always possible but in this case all of the precursors or very similar derivatives were already known.
What was their procedure?
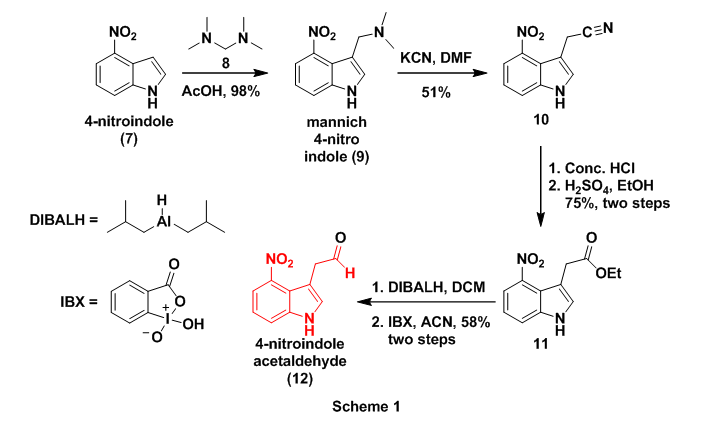
To begin, the authors prepared 4-nitroindolacetaldehyde (3) which involved a slight modification of previous reports.7 Initial work on this aldehyde commenced with the preparation of mannich derivative (9), through treatment of 4-nitroindole (7) with bis(dimethylamino)methane (8). This reaction successfully introduced one -CH2- at the 3-postion of the indole. Next dimethyl amine was replaced with a cyano(-CN) group by treatment with KCN affording cyano derivative 10. This transformation installed the second carbon on 3 which will eventually become the aldehyde carbonyl carbon. Cyano 10 then underwent acidic hydrolysis to the carboxylic acid followed by Fischer esterification with sulfuric acid and ethanol to provide ester 11. The ester was then reduced with diisobutylaluminium hydride (DIBALH) to the primary alcohol which was finally oxidized to aldehyde (3), with the hypervalent iodine compound 2-iodoxybenzoic acid (IBX), Scheme 1.
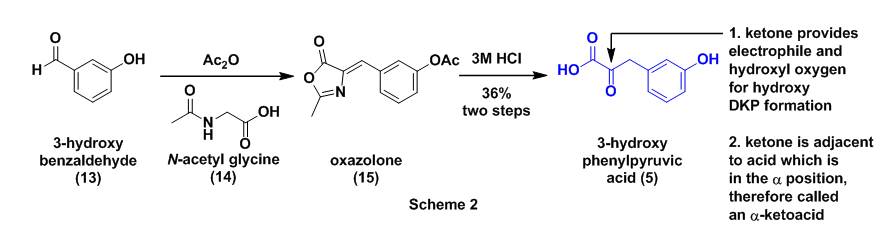
The authors next prepared 3-hydroxyphenylpyruvic acid (5) as previously described.8 The first step in this sequence involved the reaction of 3-hyroxybenzaldehyde (13) with N-acetylglycine (14) in the presence of acetic anhydride to afford the oxazolone derivative (15). Intermediate 15 then underwent acidic hydrolysis with 3M HCl to the required alpha-ketocarboxylic acid 5, Scheme 2. The key aspect to this starting material is the location of the ketone functional group alpha which is adjacent to the carboxylic acid. The presence of the ketone provides the electrophile and the hydroxyl for the final hydroxy DKP formation, see Fig. 2.
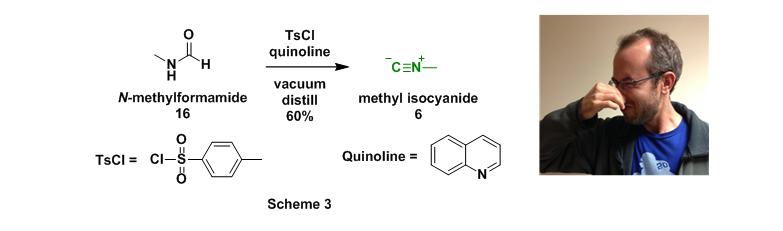
The final starting material that needed to be prepared (methyl amine, 4, is inexpensive through commercial sources) was methylisocyanide (6). This foul smelling liquid was prepared through dropwise addition of N-methylformamide (16) to p-toluenesulfonyl chloride (TsCl) dissolved in quinoline.9 The newly formed isocyanide was then simultaneously removed from the reaction mixture through vacuum distillation, Scheme 3.
With all of the required starting materials readily available the authors then conducted the all important Ugi reaction. This multicomponent coupling cascade began with the condensation of methyl amine 4 and aldehyde 12 to form imine 17. After 5 minutes isocyanide 6 and alpha-ketoacid 5 were added sequentially to the reaction mixture, which was then allowed to stir overnight at room temperature. This sequence successfully provided the desired Ugi product (2) in 37% yield. Acyclic 2 was then treated with NEt3 which provided undesired anti-1, however this compound could be converted to thaxtomin A (TA) through treatment with KOH at 70 oC, additionally 2 could be cyclized directed to TA using identical conditions, Scheme 4.
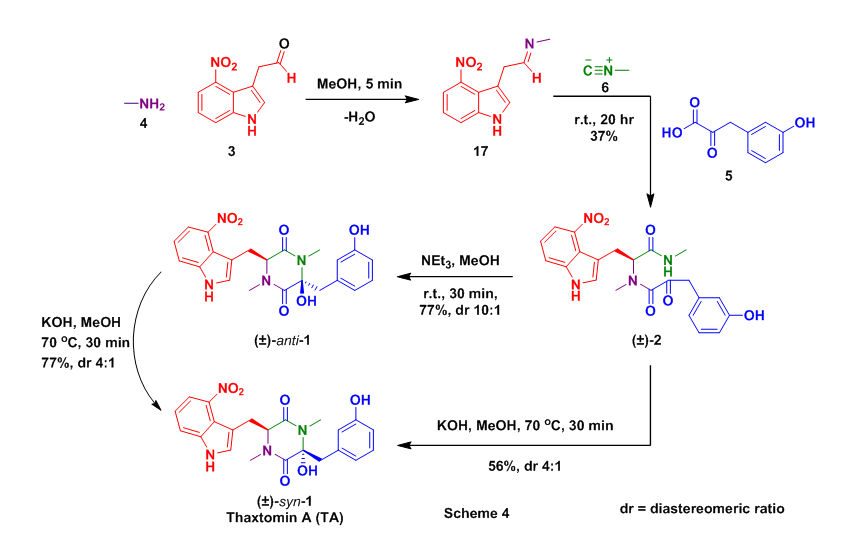
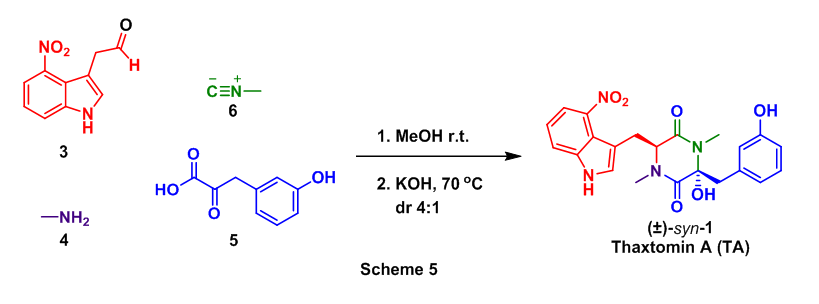
These successful step wise strategies encouraged the attempt of a one pot conversion of 3, 4, 5, and 6 to TA. In this approach the Ugi reaction was conducted as before with the addition of a cyclization step (KOH, 70 oC) after the Ugi had taken place. This tandem Ugi/cyclization sequence provided a 32% yield of TA as well as an 8% yield of anti-1, Scheme 5.
What are the conclusions?
4-nitroindolylacetaldehyde (3), methylamine (4), 3-hydroxyphenylpyruvic acid (5) and methyl isocyanide (6) were used in a tandem one-pot Ugi/DKP cyclization process for the natural product thaxtomin A. While this synthesis is inherently limited by the Ugi reaction itself in that only racemic TA is obtained, it clearly demonstrates the power of multicomponent reactions to generate biologically validated scaffolds. Including those embedded in naturally occurring products, in a limited number of synthetic steps in the absence of protecting groups.
What are the next steps?
The authors plan to use the Ugi/cylization sequence described to prepare a library of hydroxy DKP derivatives which will be studied as potential herbicides.
NMR spectroscopy data for (±)-thaxtomin A
1 H NMR data for TA, syn-1
The1H NMR spectra were recorded with a Bruker MHz 600 spectrometer for TA, syn-1. Deuterated methanol (CD3OD) was used as a solvent.
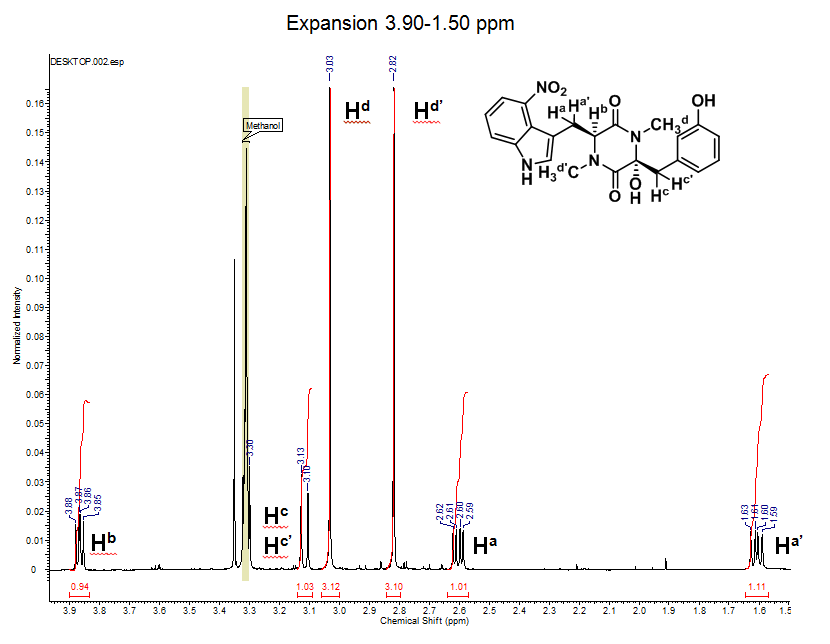
1H NMR (600 MHz, CD3OD): d 7.84 (dd, J = 7.9 Hz, 1H), 7.69 (d, J = 8.0 Hz, 1H), 7.23 (t, J = 8.1 Hz, 1H), 7.18 (t, J = 8.0 Hz, 1H), 6.95 (s, 1H), 6.75 (m, 1H), 6.70 (m, 2H), 3.86 (dd, J1 = 6.4 Hz, J2 = 8.8 Hz, 1H), 3.31 (overlapping with solvent residual peak, 1H), 3.13 (d, J = 13.5 Hz, 1H), 3.03 (s, 3H), 2.82 (s, 3H), 2.60 (dd, J1 = 6.3 Hz, J2 = 14.1 Hz, 1H), 1.61 (dd, J1 = 8.9 Hz, J2 = 14.1 Hz, 1H)
Explanation
- The NMR peaks are labeled according to the structure of thaxtomin A, while exactly how the hydrogens were labeled may be beyond the scope of this exercise. It is important to keep in mind that each signal in a 1H NMR spectrum represents distinct hydrogen(s) present on the molecule undergoing the experiment.
- Values found directly above the peaks (blue) are related to how much electron density is surrounding the proton, higher number (downfield) means less electron density and lower number (upfield) means more electron density. These values are called the chemical shift of the hydrogen(s). Unfortunately Hc’s chemical shift is identical to the solvent residual peak of deuterated methanol (3.31), but if you look closely the edges of the Hc signal can be seen.
- The integration values can be found directly under each peak (# in red), these values represent the number of hydrogen atoms associated with the NMR signal.
- For example the peak at 3.03 ppm has an integration ratio right around 3, which makes sense because this singlet represents a methyl group that contains 3 hydrogens.
- Integration ratios found in routine NMR seldom give the exact value but are usually within 10% of the actual number of hydrogen(s), therefore rounding to the nearest whole number is common.
Explanation
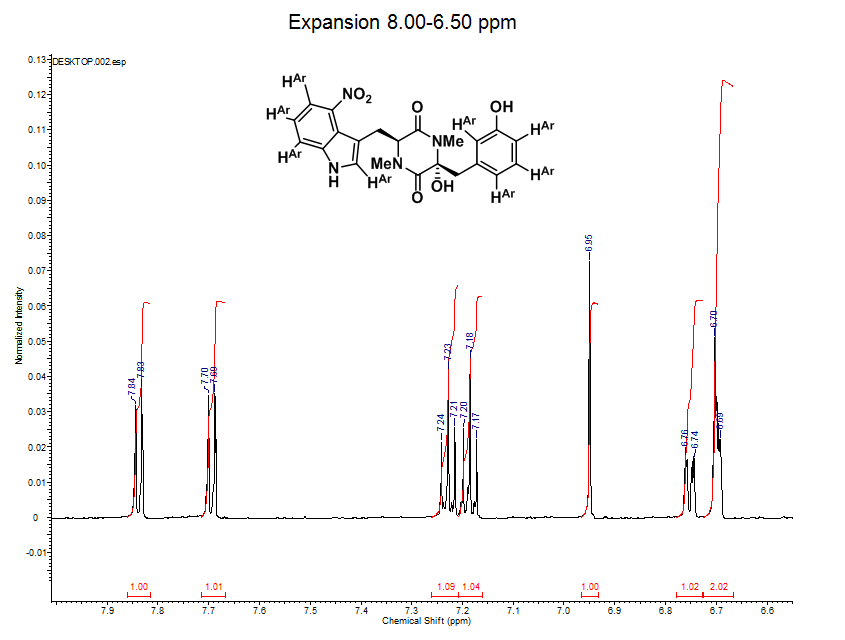
- This expansion represents the 8 aromatic hydrogens on thaxtomin A (labeled has HAr on the structure).
13C NMR for TA, syn-1
The 13C NMR spectra were recorded with a Bruker 150 MHz spectrometer for TA, syn-1. Deuterated methanol (CD3OD) was used as a solvent.
13C NMR (150 MHz, CD3OD): d 168.3, 166.8, 159.1, 143.6, 141.1, 137.4, 132.5, 131.2, 122.7, 121.0, 119.8, 119.3, 118.6, 118.4, 115.9, 110.5, 88.0, 64.6, 43.5, 34.2, 33.5, 28.5
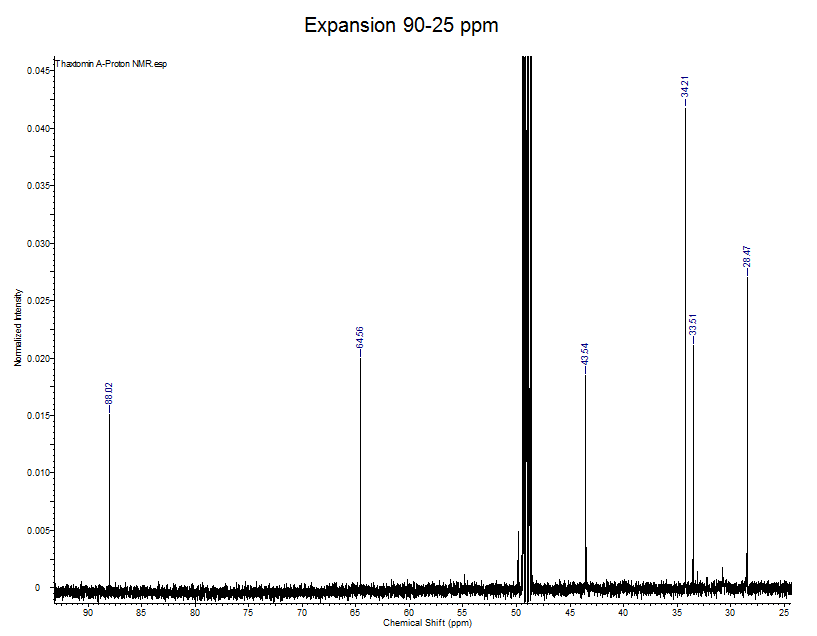
Explanation
- Carbon NMR (0-200 ppm) has a much larger scale then proton NMR (0-10 ppm).
- The expanded spectrum above displays all of the aliphatic (non-aromatic) C atoms present in thaxtomin A.
- There are 6 lines in this expansion and there are 6 aliphatic Cs on thaxtomin A, can you find the aliphatic carbons on the structure?
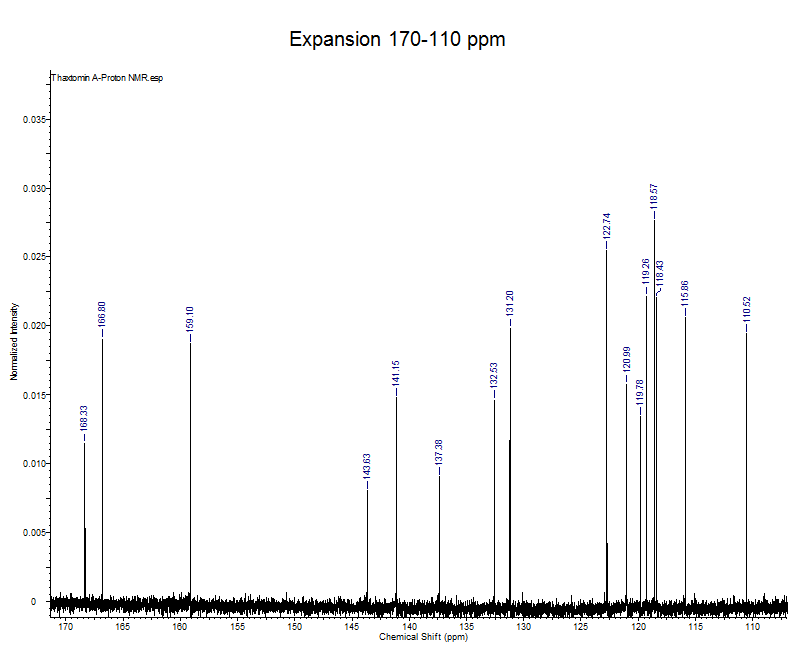
Explanation
- This expansion represents all the aromatic and amide carbonyl signals.
- The amide carbonyl signals are the furthest downfield at 166.80 and 168.33.
- The aromatic signals of thaxtomin A can be found between 160-110 ppm, because there is no symmetry in the aromatic rings all 14 aromatic carbons can be accounted for.
Abbreviations used in text
Abbreviation | Name | Role | Links |
DIBALH | Diisobutylaluminium hydride | Reducing agent for the conversion of the ester to a primary alcohol | |
DKP | 2,5-diketopiperazine | Class of cyclic dipetides | |
DOS | Diversity-Oriented Synthesis | A strategy for quick access to molecule libraries with an emphasis on skeletal diversity | |
HCl | Hydrochloric acid | Acid used as an aqueous source of H+ for the hydrolysis of the cyano group to a carboxylic acid | |
IBX | 2-iodoxybenzoic acid | Oxidizing agent used to convert the primary alcohol to an aldehyde | |
KCN | Potassium cyanide | Nucleophile used to add an additional carbon atom | |
KOH | Potassium hydroxide | Base used in the cylization of the acylic Ugi intermediate | |
TA | Thaxtomin A | Natural bacterial herbicide/synthetic target | |
TsCl | P-toluenesulfonyl chloride | Sulfonating reagent, which turns a free OH into a leaving group |
References
1. Gianessi, L. P.; Reigner, N. P. Weed Technology 2007, 21, 559-566.
2. Senseman, S. A.; Grey, T. L. Weed Science 2013, 62, 382-384.
3. King, R. R.; Lawrence, C. H.; Clark, M. C.; Calhoun, L. A. J. Chem. Soc., Chem. Commun. 1989, 849-850.
4. (a) Koivunen, M.; Marrone, P.; Boddy, L. Uses of thaxtomin and thaxtomin compositions as herbicides. WO2014013343A2, 2014. (b) Inman, S.; Semones, S. Methods of controlling weeds with thaxtomin and thaxtomin compositions in combination with a beneficial herbicide. WO2013066894A2, 2013. (c) Leep, D.; Doricchi, L.; Perez Baz, M. J.; Millan, F. R.; Fernandez Chimeno, R. I. Use of thaxtomin for selective control of rice and aquatic based weeds. WO2010121079A2, 2010.
5. King, R. R.; Calhoun, L. A. Phytochemistry (Elsevier) 2009, 70, 833-841.
6. Zhang, H.; Ning, X.; Hang, H.; Ru, X.; Li, H.; Li, Y.; Wang, L.; Zhang, X.; Yu, S.; Qiao, Y.; Wang, X.; Wang, P. G. Org. Lett. 2013.
7. (a) Revelant, G.; Dunand, S.; Hesse, S. p.; Kirsch, G. Synthesis 2011, 2011, 2935-2940. (b) Bell, I.; Stump, C. A. Preparation of spirolactam-containing tricyclic aryl and heteroaryl amides as CGRP receptor antagonists for the treatment of headaches, including migraine and cluster headaches. WO2008153852A1, 2008.
8. Wong, H. N. C.; Xu, Z. L.; Chang, H. M.; Lee, C. M. Synthesis 1992, 1992, 793-797.
9. Schuster, R. E. S., J. E.; Casanova, J. Organic Syntheses 1966, 46, 75.
Additional information
Journal articles made easy are journal articles from a range of Royal Society of Chemistry journals that have been re-written into a standard, accessible format. They contain links to ChemSpider entries, related journal articles, books and Learn Chemistry resources such as videos of techniques, and resources on theory and activities. They should facilitate students understanding of scientific journal articles and how to extract and interpret the information in them.





No comments yet